

Discrete modeling for integration and analysis of large-scale signaling networks
- PMID: 35696426
- PMCID: PMC9232147
- DOI: 10.1371/journal.pcbi.1010175
Discrete modeling for integration and analysis of large-scale signaling networks
Abstract
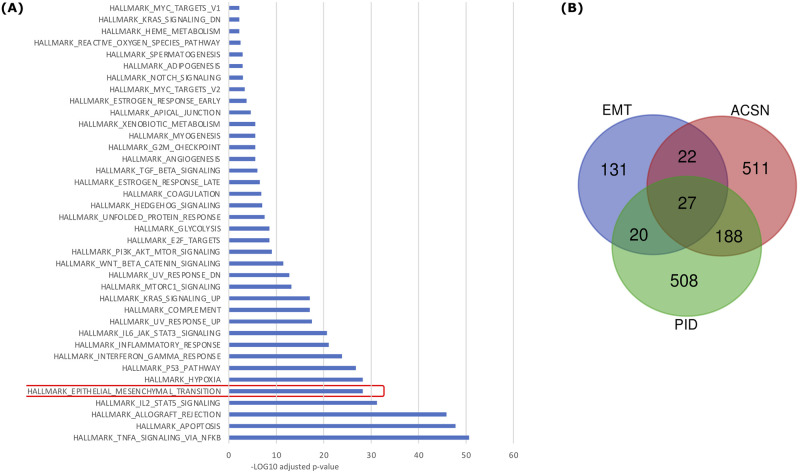
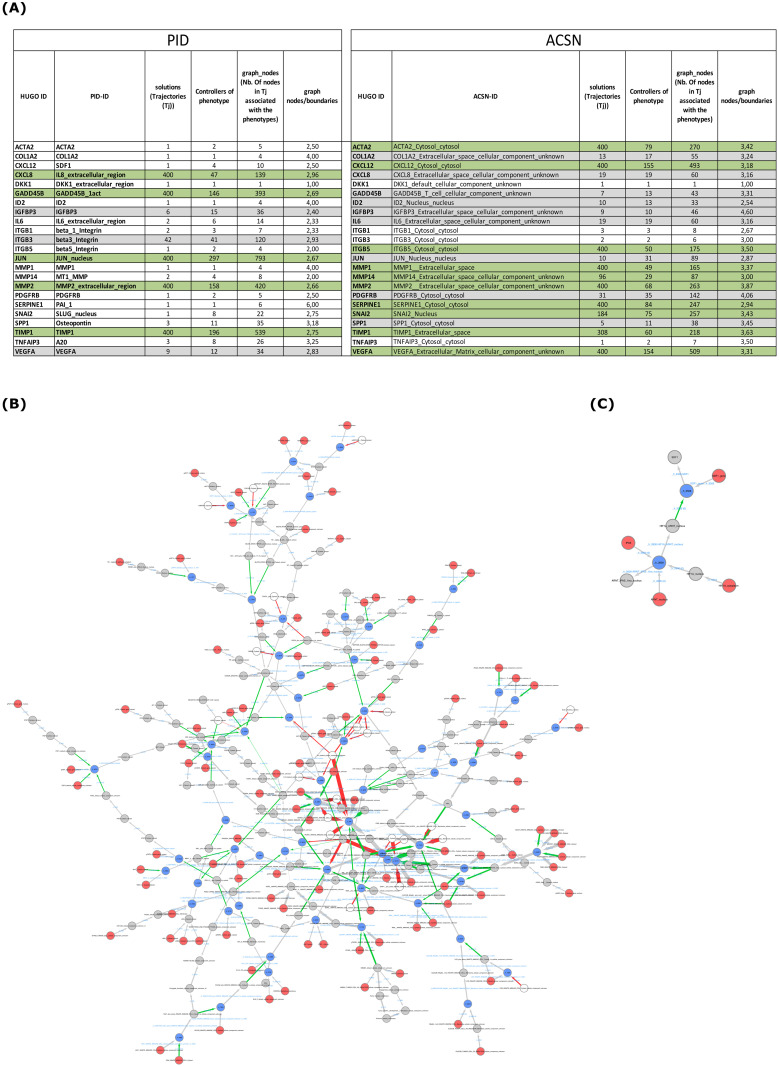
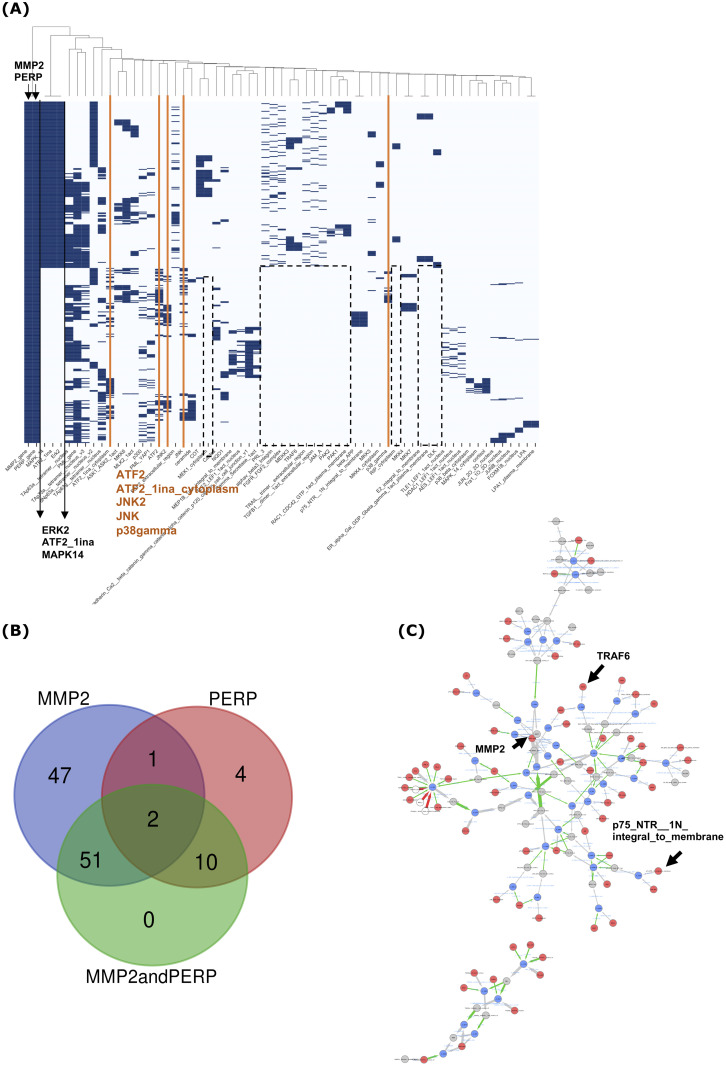
Most biological processes are orchestrated by large-scale molecular networks which are described in large-scale model repositories and whose dynamics are extremely complex. An observed phenotype is a state of this system that results from control mechanisms whose identification is key to its understanding. The Biological Pathway Exchange (BioPAX) format is widely used to standardize the biological information relative to regulatory processes. However, few modeling approaches developed so far enable for computing the events that control a phenotype in large-scale networks. Here we developed an integrated approach to build large-scale dynamic networks from BioPAX knowledge databases in order to analyse trajectories and to identify sets of biological entities that control a phenotype. The Cadbiom approach relies on the guarded transitions formalism, a discrete modeling approach which models a system dynamics by taking into account competition and cooperation events in chains of reactions. The method can be applied to every BioPAX (large-scale) model thanks to a specific package which automatically generates Cadbiom models from BioPAX files. The Cadbiom framework was applied to the BioPAX version of two resources (PID, KEGG) of the Pathway Commons database and to the Atlas of Cancer Signalling Network (ACSN). As a case-study, it was used to characterize sets of biological entities implicated in the epithelial-mesenchymal transition. Our results highlight the similarities between the PID and ACSN resources in terms of biological content, and underline the heterogeneity of usage of the BioPAX semantics limiting the fusion of models that require curation. Causality analyses demonstrate the smart complementarity of the databases in terms of combinatorics of controllers that explain a phenotype. From a biological perspective, our results show the specificity of controllers for epithelial and mesenchymal phenotypes that are consistent with the literature and identify a novel signature for intermediate states.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures





References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
