

Cell deaths: Involvement in the pathogenesis and intervention therapy of COVID-19
- PMID: 35697684
- PMCID: PMC9189267
- DOI: 10.1038/s41392-022-01043-6
Cell deaths: Involvement in the pathogenesis and intervention therapy of COVID-19
Abstract
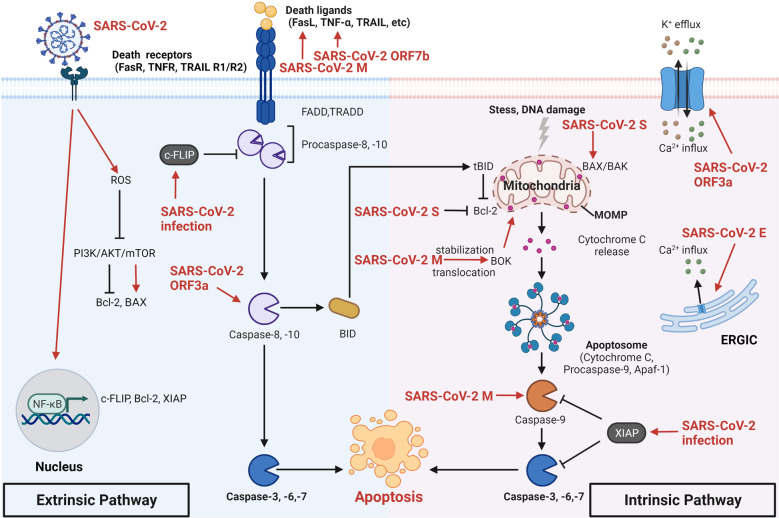
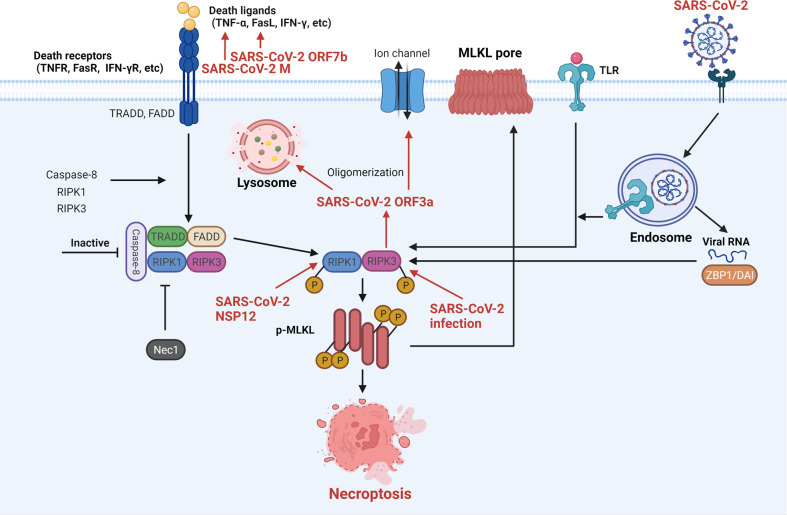
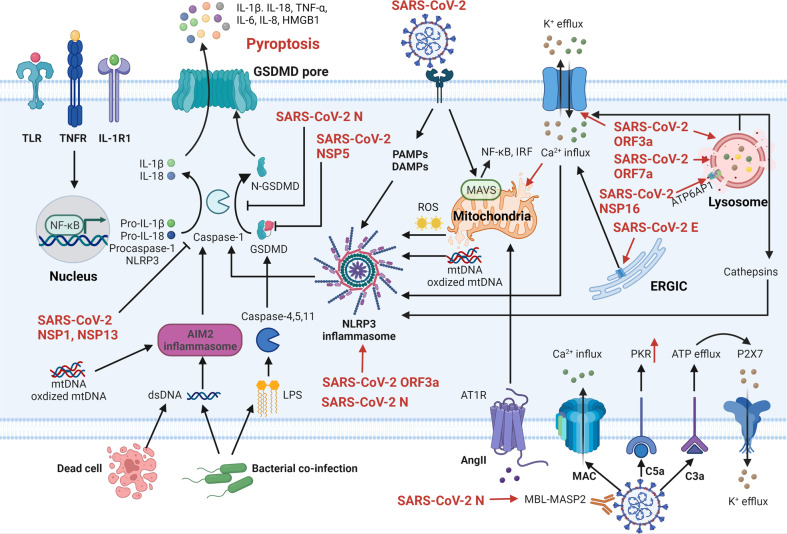
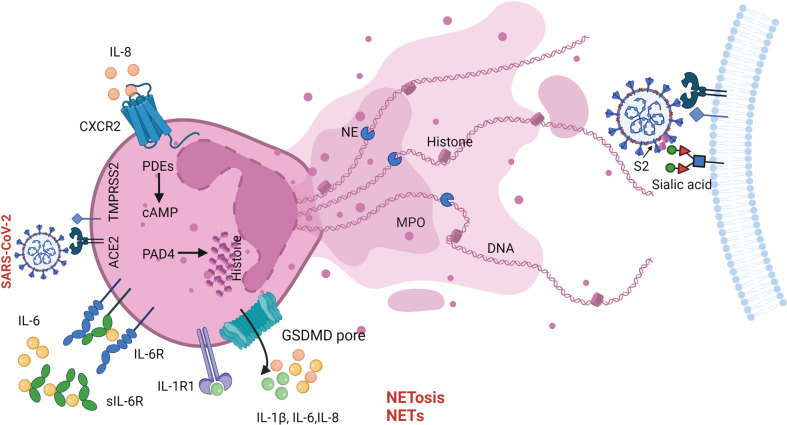
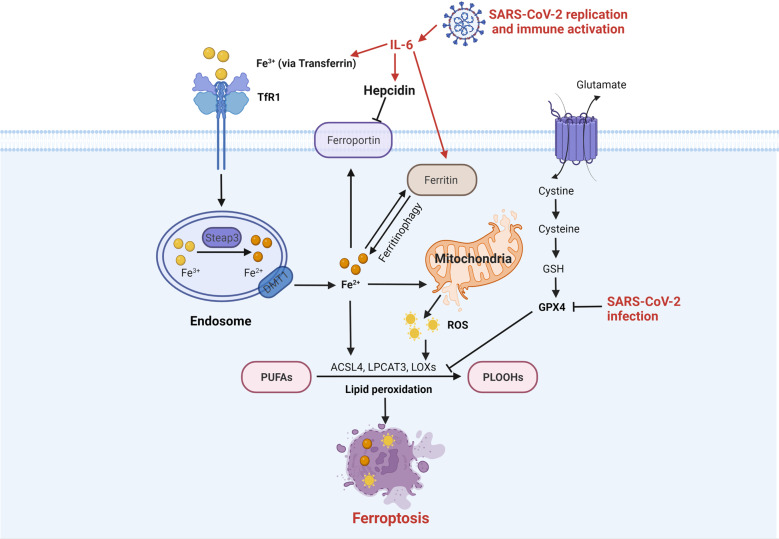
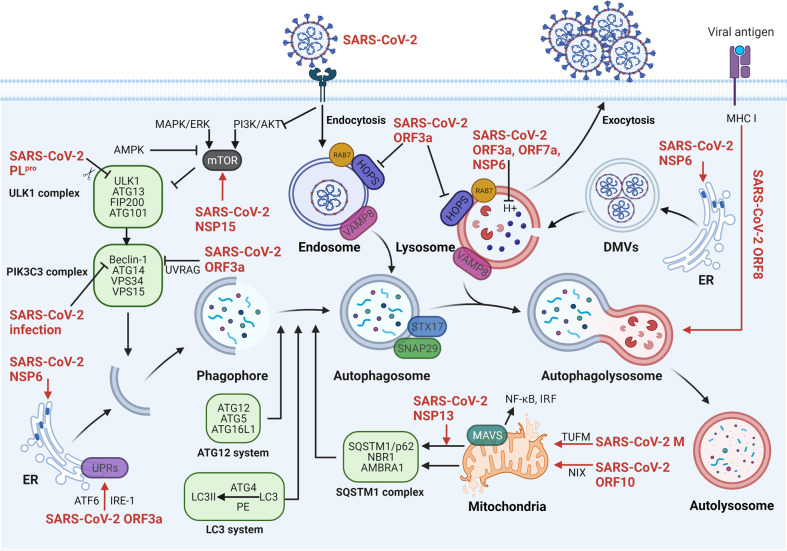
The current pandemic of coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection has dramatically influenced various aspects of the world. It is urgent to thoroughly study pathology and underlying mechanisms for developing effective strategies to prevent and treat this threatening disease. It is universally acknowledged that cell death and cell autophagy are essential and crucial to maintaining host homeostasis and participating in disease pathogenesis. At present, more than twenty different types of cell death have been discovered, some parts of which have been fully understood, whereas some of which need more investigation. Increasing studies have indicated that cell death and cell autophagy caused by coronavirus might play an important role in virus infection and pathogenicity. However, the knowledge of the interactions and related mechanisms of SARS-CoV-2 between cell death and cell autophagy lacks systematic elucidation. Therefore, in this review, we comprehensively delineate how SARS-CoV-2 manipulates diverse cell death (including apoptosis, necroptosis, pyroptosis, ferroptosis, and NETosis) and cell autophagy for itself benefits, which is simultaneously involved in the occurrence and progression of COVID-19, aiming to provide a reasonable basis for the existing interventions and further development of novel therapies.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
The role of reactive oxygen species in severe acute respiratory syndrome coronavirus 2 (SARS-COV-2) infection-induced cell death.Cell Mol Biol Lett. 2024 Nov 8;29(1):138. doi: 10.1186/s11658-024-00659-6. Cell Mol Biol Lett. 2024. PMID: 39516736 Free PMC article. Review.
-
The role of cell death in SARS-CoV-2 infection.Signal Transduct Target Ther. 2023 Sep 20;8(1):357. doi: 10.1038/s41392-023-01580-8. Signal Transduct Target Ther. 2023. PMID: 37726282 Free PMC article. Review.
-
Coronavirus Infection-Associated Cell Death Signaling and Potential Therapeutic Targets.Molecules. 2021 Dec 9;26(24):7459. doi: 10.3390/molecules26247459. Molecules. 2021. PMID: 34946543 Free PMC article. Review.
-
Therapy Targets SARS-CoV-2 Infection-Induced Cell Death.Front Immunol. 2022 May 17;13:870216. doi: 10.3389/fimmu.2022.870216. eCollection 2022. Front Immunol. 2022. PMID: 35655782 Free PMC article. Review.
-
The roles of cellular protease interactions in viral infections and programmed cell death: a lesson learned from the SARS-CoV-2 outbreak and COVID-19 pandemic.Pharmacol Rep. 2022 Dec;74(6):1149-1165. doi: 10.1007/s43440-022-00394-9. Epub 2022 Aug 23. Pharmacol Rep. 2022. PMID: 35997950 Free PMC article. Review.
Cited by
-
Identification of FasL as a crucial host factor driving COVID-19 pathology and lethality.Cell Death Differ. 2024 May;31(5):544-557. doi: 10.1038/s41418-024-01278-6. Epub 2024 Mar 21. Cell Death Differ. 2024. PMID: 38514848 Free PMC article.
-
Pharmacogenomic Landscape of Ivermectin and Selective Antioxidants: Exploring Gene Interplay in the Context of Long COVID.Int J Mol Sci. 2023 Oct 23;24(20):15471. doi: 10.3390/ijms242015471. Int J Mol Sci. 2023. PMID: 37895148 Free PMC article.
-
Alveolar macrophages: Achilles' heel of SARS-CoV-2 infection.Signal Transduct Target Ther. 2022 Jul 19;7(1):242. doi: 10.1038/s41392-022-01106-8. Signal Transduct Target Ther. 2022. PMID: 35853858 Free PMC article. Review.
-
Innate immunity, therapeutic targets and monoclonal antibodies in SARS-CoV-2 infection.PeerJ. 2025 Jun 20;13:e19462. doi: 10.7717/peerj.19462. eCollection 2025. PeerJ. 2025. PMID: 40552037 Free PMC article. Review.
-
Cerebral small vessel injury in mice with damage to ACE2-expressing cerebral vascular endothelial cells and post COVID-19 patients.Alzheimers Dement. 2024 Nov;20(11):7971-7988. doi: 10.1002/alz.14279. Epub 2024 Oct 1. Alzheimers Dement. 2024. PMID: 39352003 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
