

Integrated multi-omics reveal polycomb repressive complex 2 restricts human trophoblast induction
- PMID: 35697783
- PMCID: PMC9203278
- DOI: 10.1038/s41556-022-00932-w
Integrated multi-omics reveal polycomb repressive complex 2 restricts human trophoblast induction
Abstract
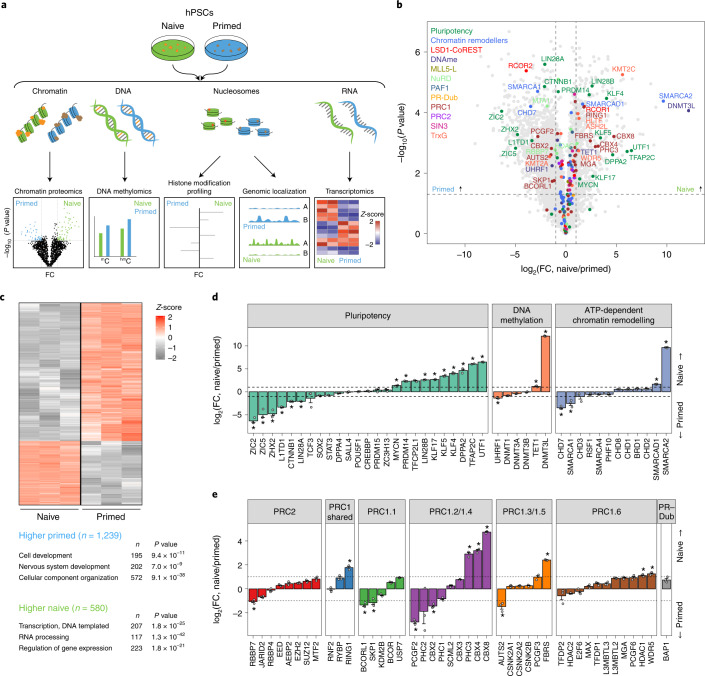
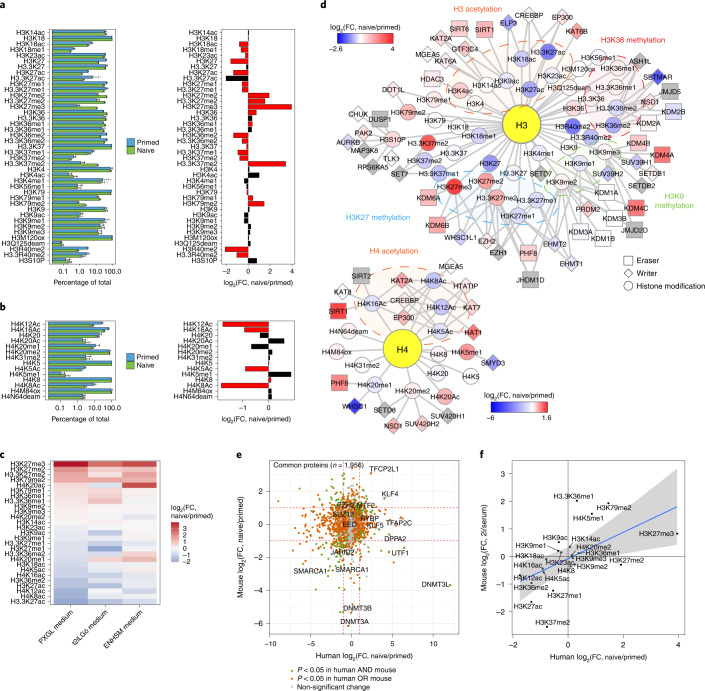
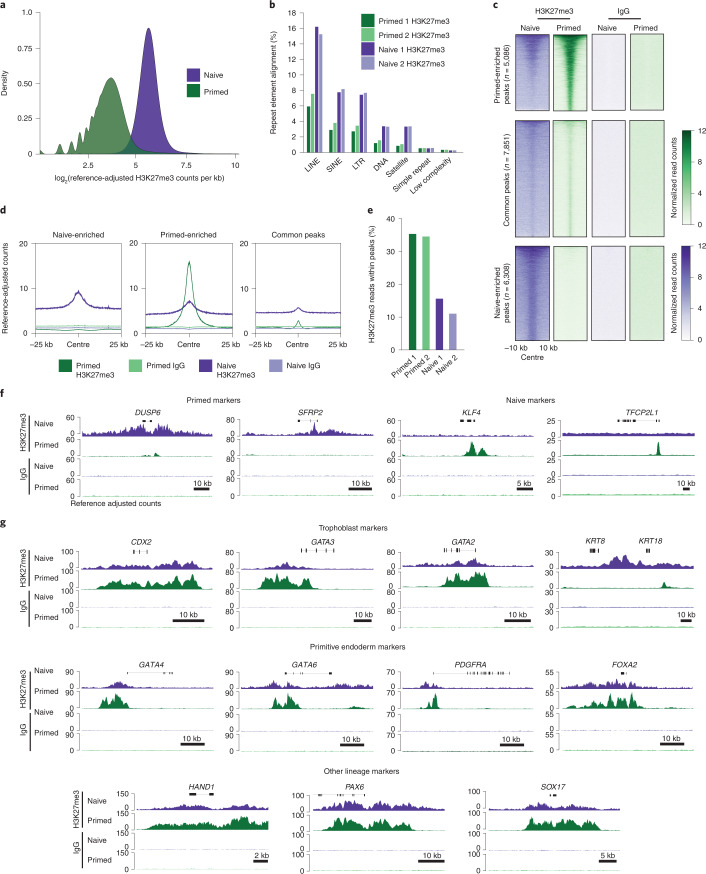
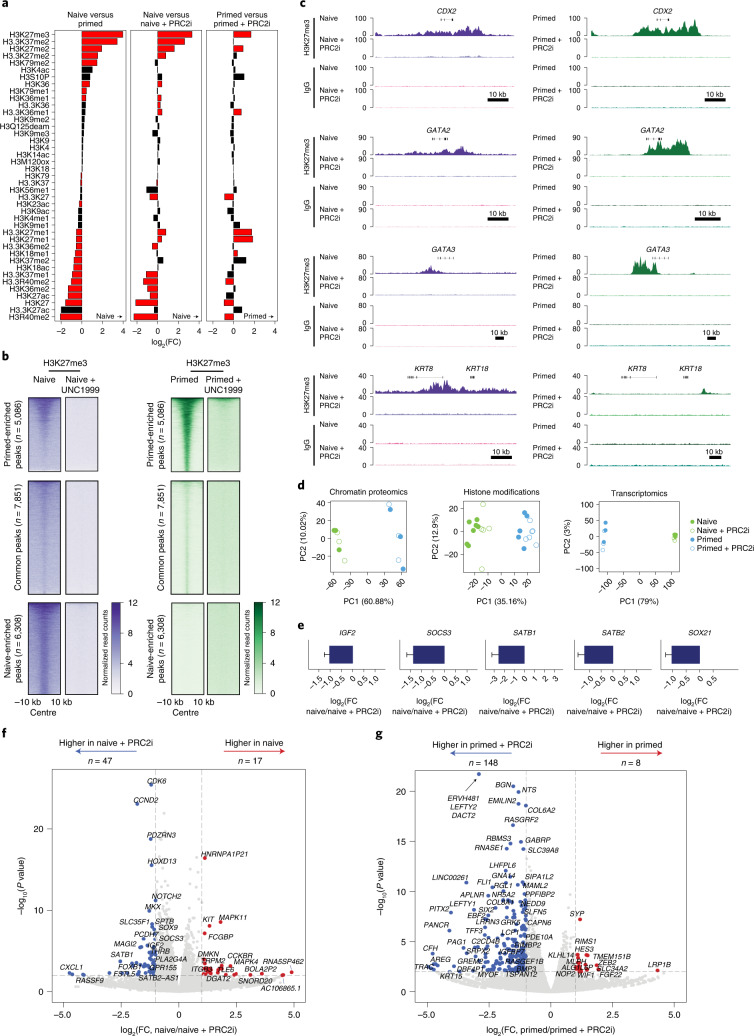
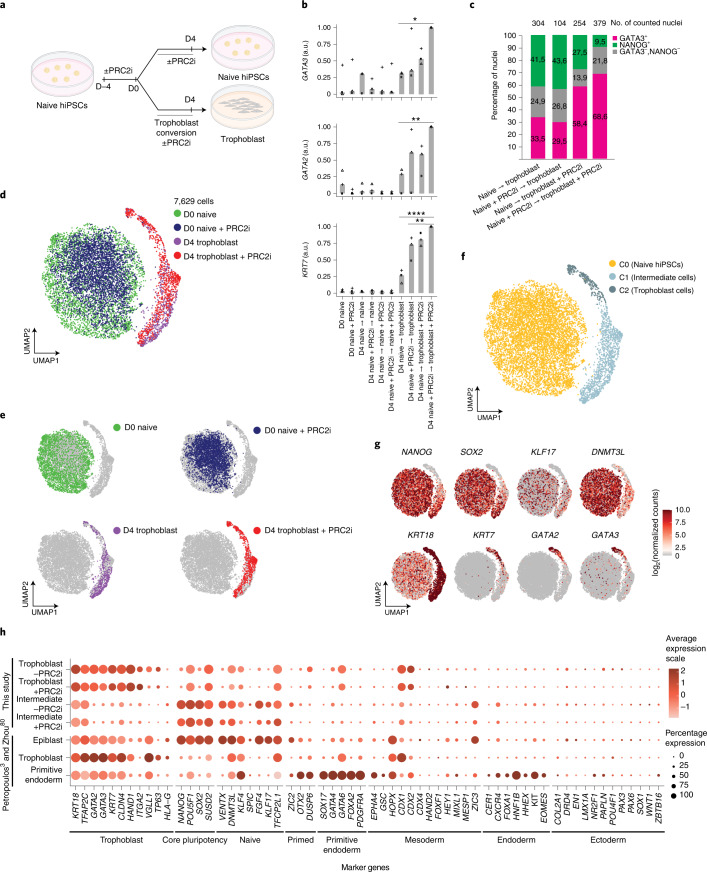
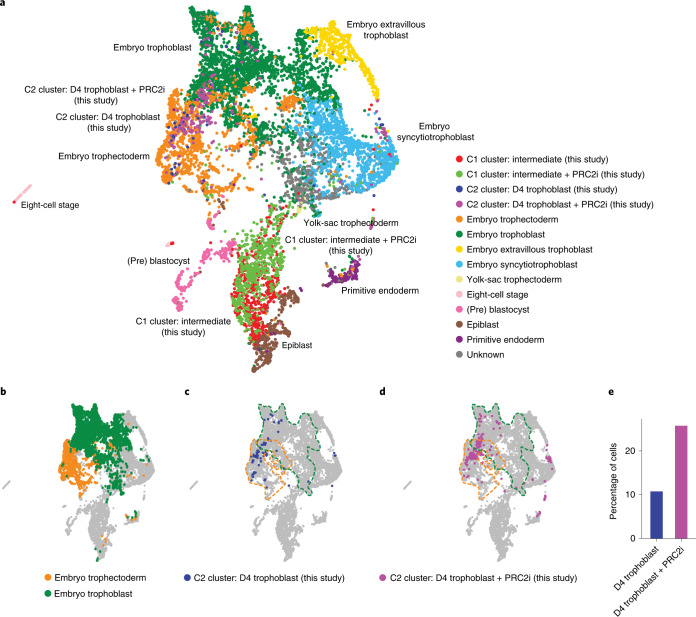
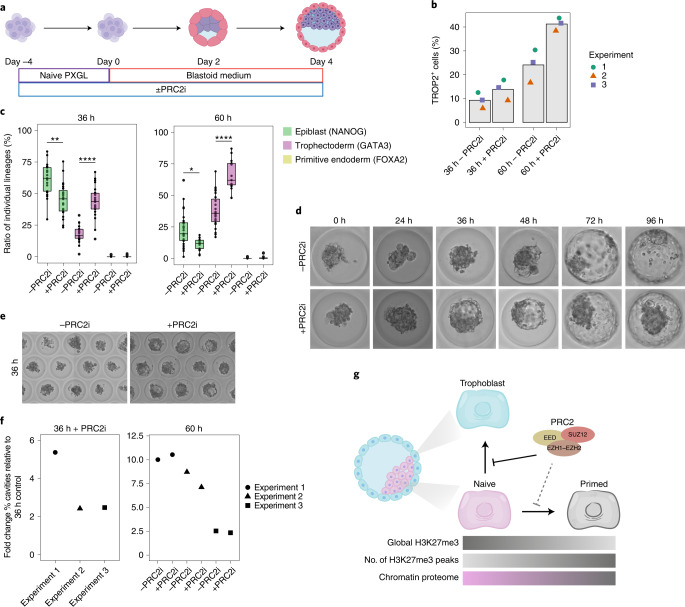
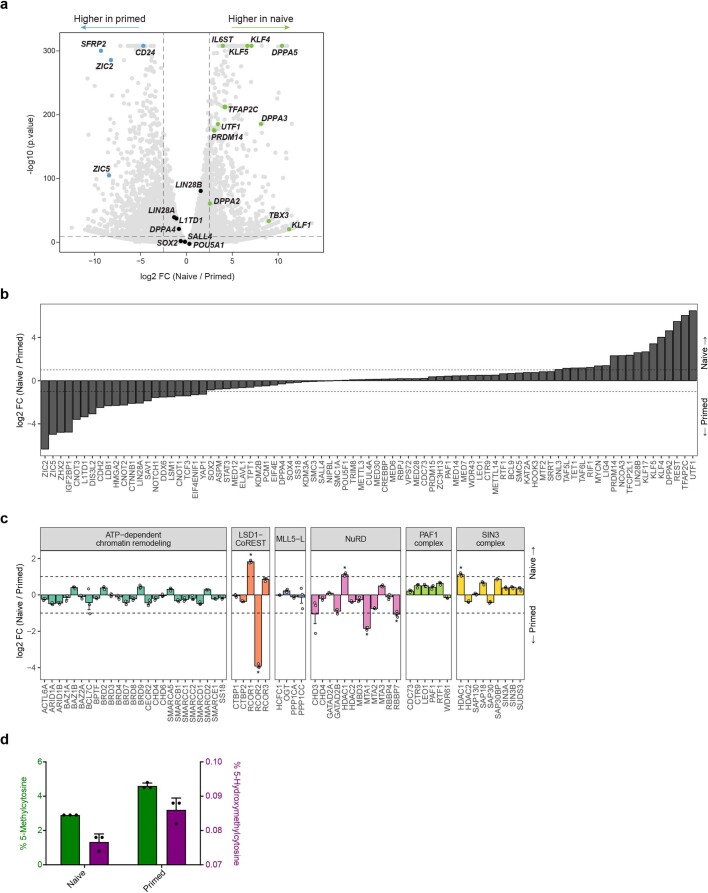
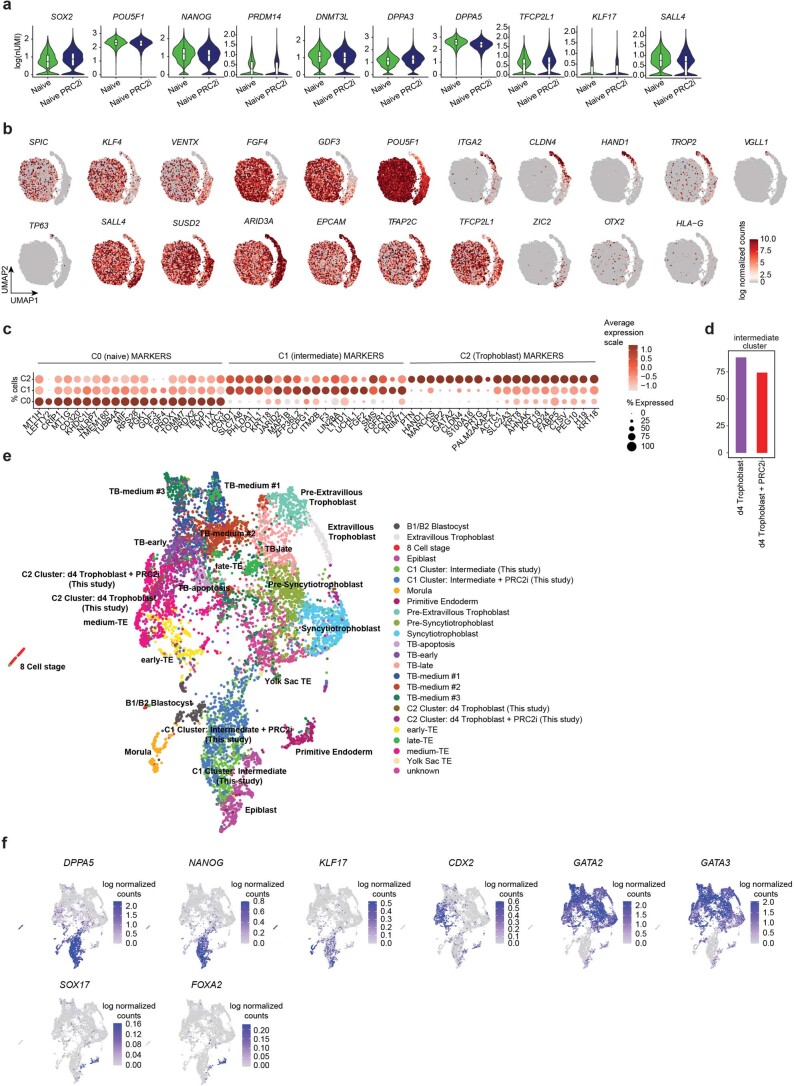
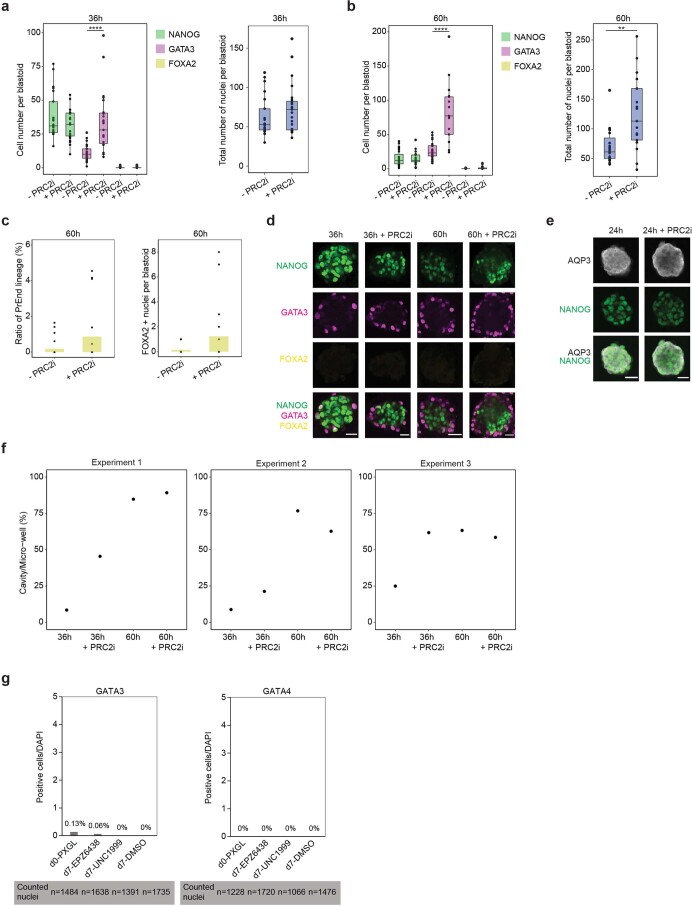
Human naive pluripotent stem cells have unrestricted lineage potential. Underpinning this property, naive cells are thought to lack chromatin-based lineage barriers. However, this assumption has not been tested. Here we define the chromatin-associated proteome, histone post-translational modifications and transcriptome of human naive and primed pluripotent stem cells. Our integrated analysis reveals differences in the relative abundance and activities of distinct chromatin modules. We identify a strong enrichment of polycomb repressive complex 2 (PRC2)-associated H3K27me3 in the chromatin of naive pluripotent stem cells and H3K27me3 enrichment at promoters of lineage-determining genes, including trophoblast regulators. PRC2 activity acts as a chromatin barrier restricting the differentiation of naive cells towards the trophoblast lineage, whereas inhibition of PRC2 promotes trophoblast-fate induction and cavity formation in human blastoids. Together, our results establish that human naive pluripotent stem cells are not epigenetically unrestricted, but instead possess chromatin mechanisms that oppose the induction of alternative cell fates.
© 2022. The Author(s).
Conflict of interest statement
The Institute for Molecular Biotechnology, Austrian Academy of Sciences has filed patent application no. EP21151455.9 describing the protocols for human blastoid formation and for the blastoid–endometrium interaction assay. A.J. and N.R. are the inventors on this patent. All other authors declare no competing interests.
Figures
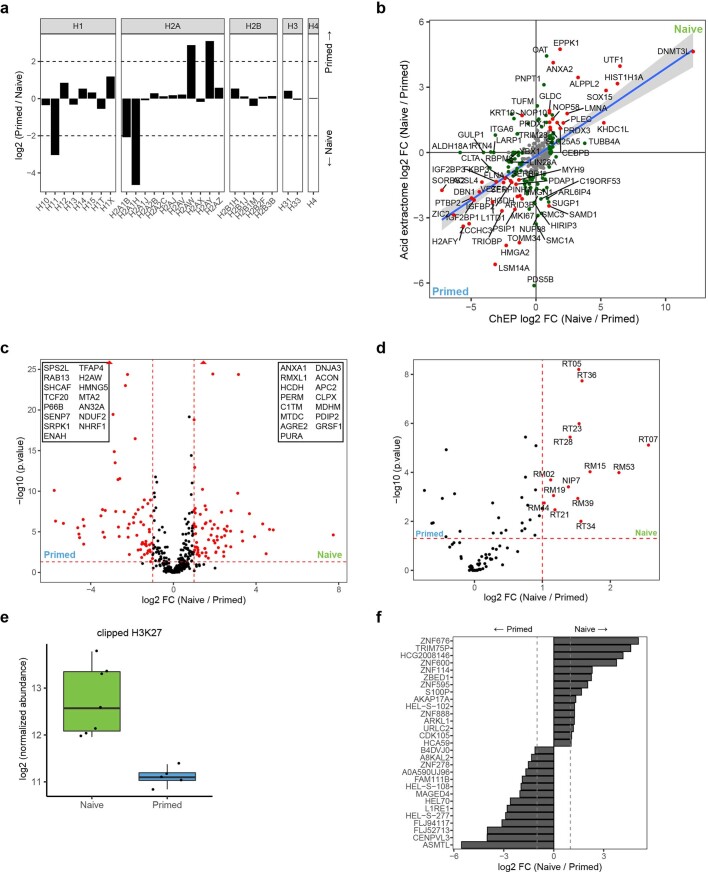
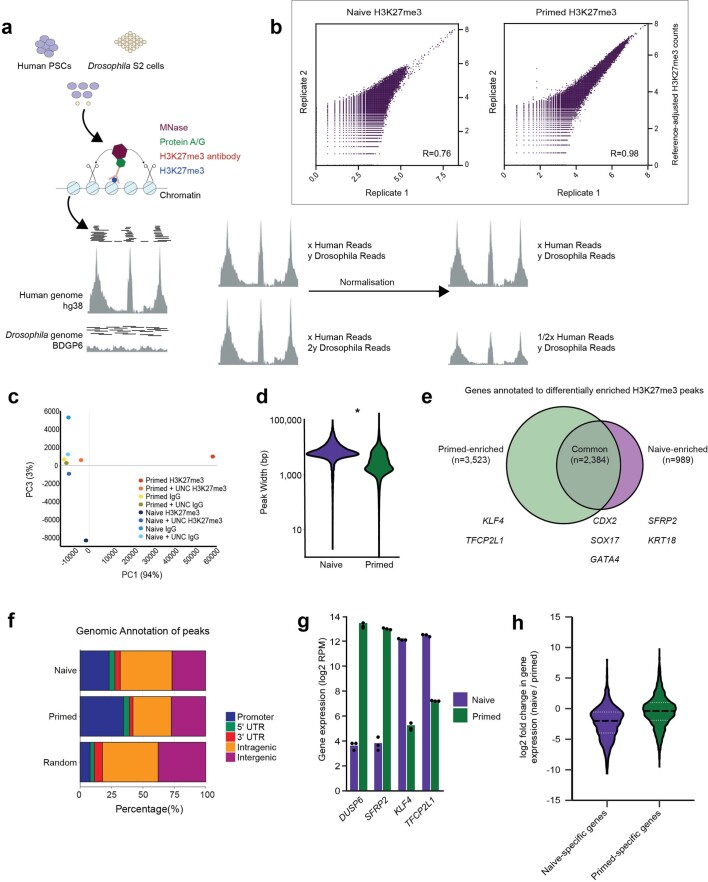
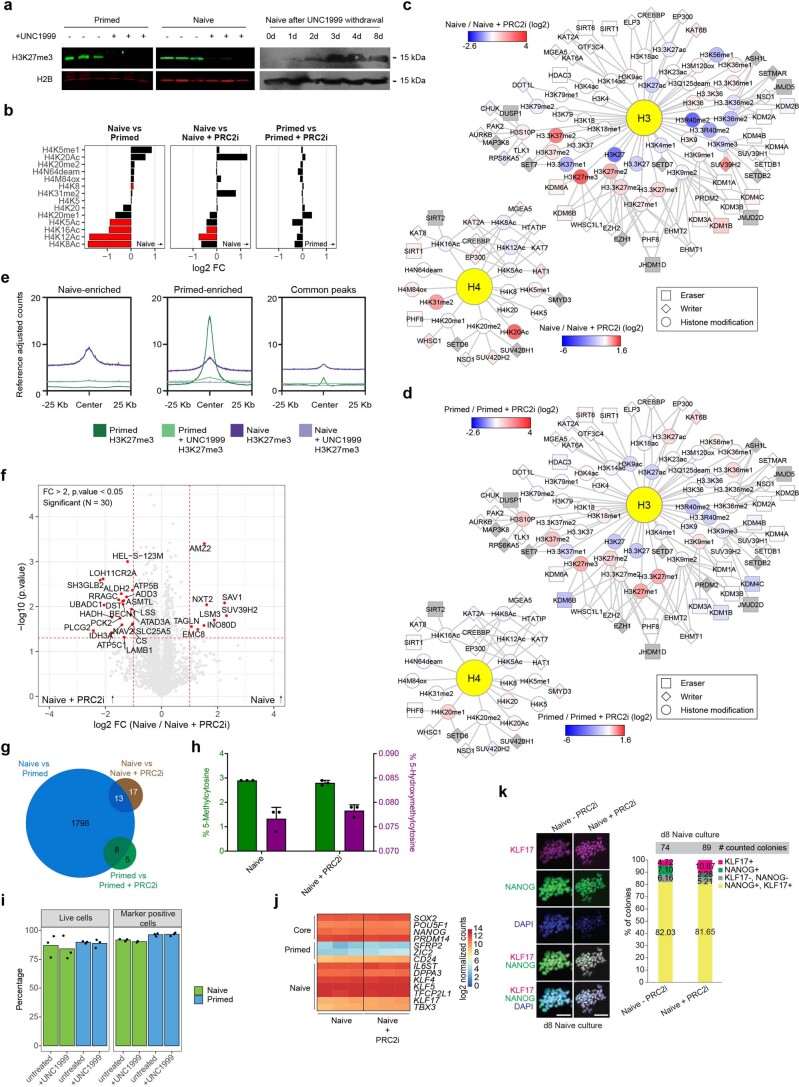
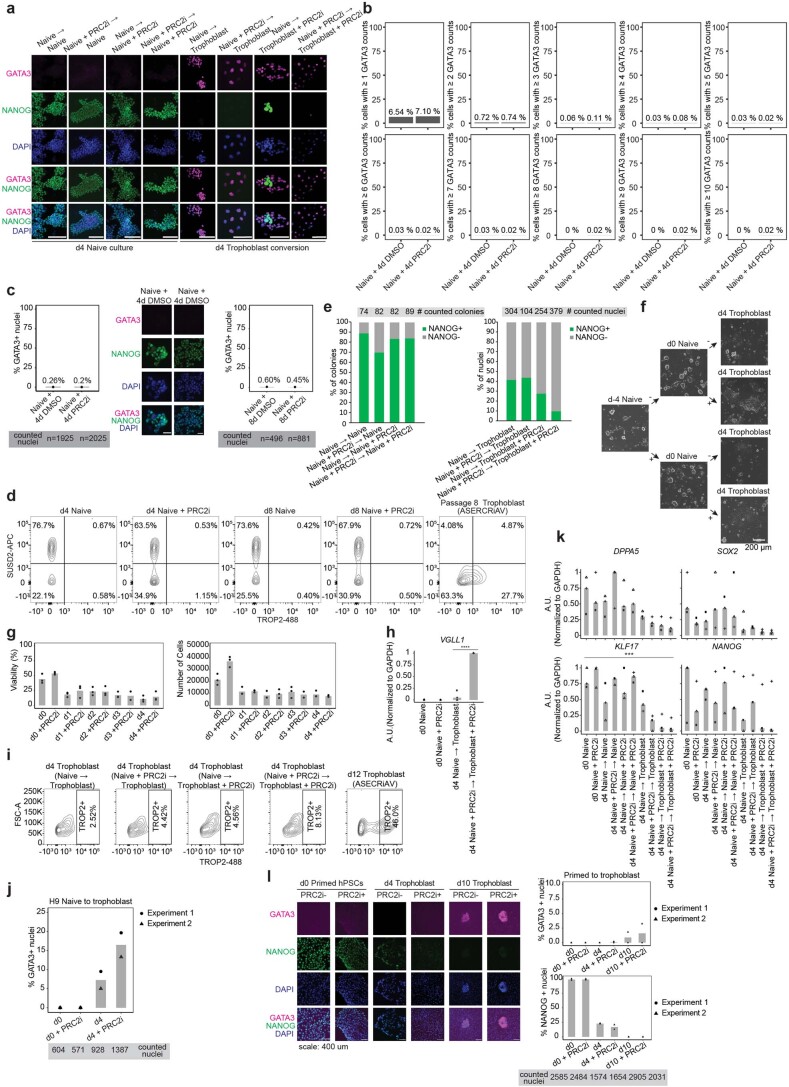
Comment in
-
PRC2 shields the potency of human stem cells.Nat Cell Biol. 2022 Jun;24(6):806-808. doi: 10.1038/s41556-022-00937-5. Nat Cell Biol. 2022. PMID: 35697784 No abstract available.
References
-
- Rossant J. Investigation of the determinative state of the mouse inner cell mass. II. The fate of isolated inner cell masses transferred to the oviduct. J. Embryol. Exp. Morphol. 1975;33:991–1001. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 102160/Z/13/Z/WT_/Wellcome Trust/United Kingdom
- P01 CA196539/CA/NCI NIH HHS/United States
- BBS/E/B/000C0421/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- P01 AG031862/AG/NIA NIH HHS/United States
- 215116/Z/18/Z/WT_/Wellcome Trust/United Kingdom
- BBS/E/B/000C0422/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- R01 AI118891/AI/NIAID NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- MR/N018419/1/MRC_/Medical Research Council/United Kingdom
- MR/V02969X/1/MRC_/Medical Research Council/United Kingdom
- MR/J003808/1/MRC_/Medical Research Council/United Kingdom
- MR/T011769/1/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
