

Estimation of tumor cell total mRNA expression in 15 cancer types predicts disease progression
- PMID: 35697807
- PMCID: PMC9646498
- DOI: 10.1038/s41587-022-01342-x
Estimation of tumor cell total mRNA expression in 15 cancer types predicts disease progression
Abstract
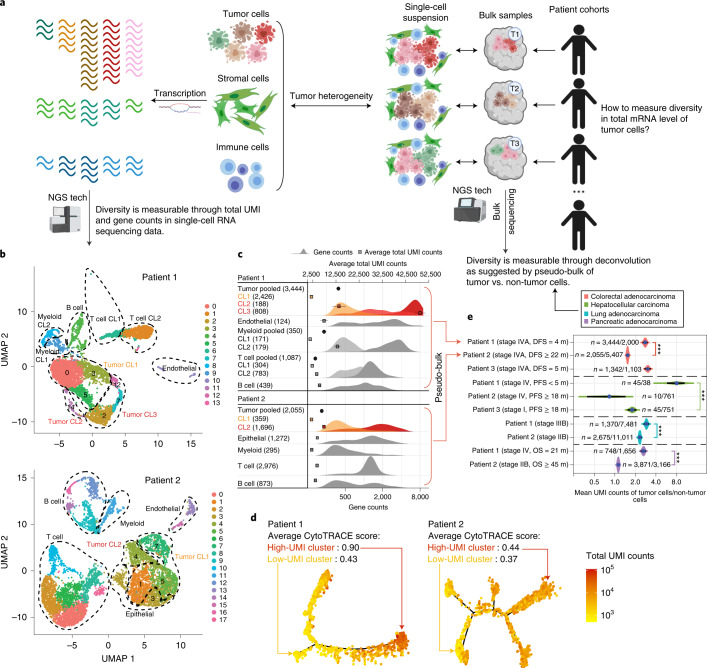
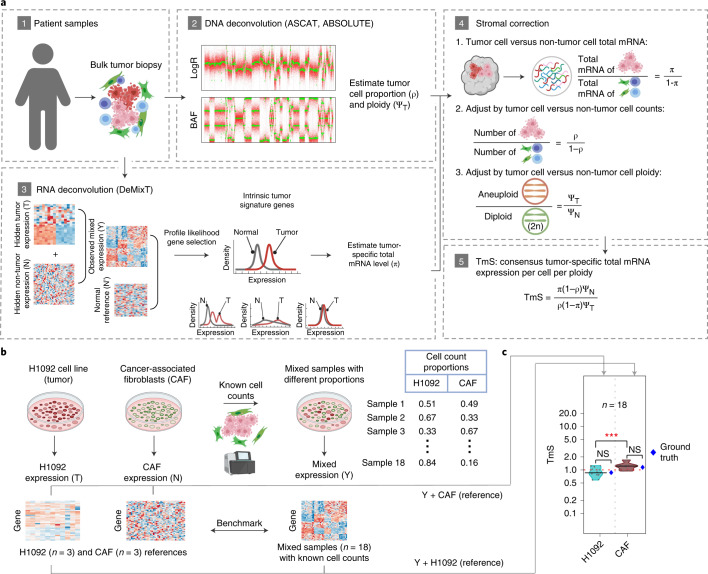
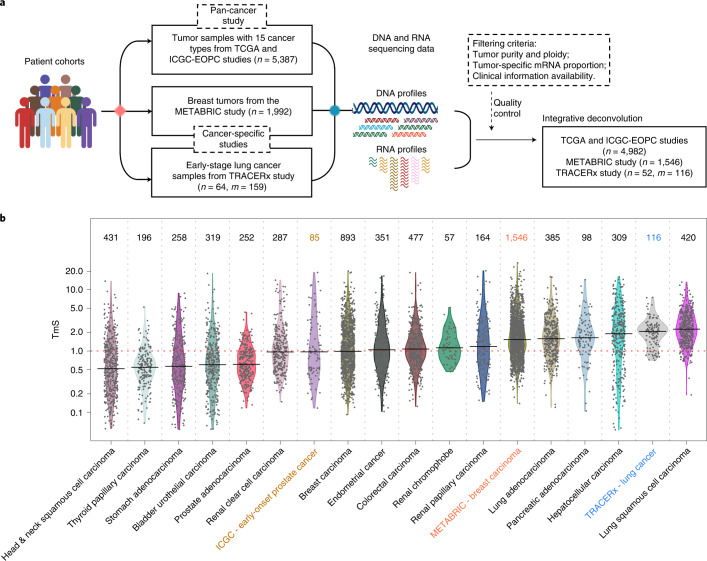
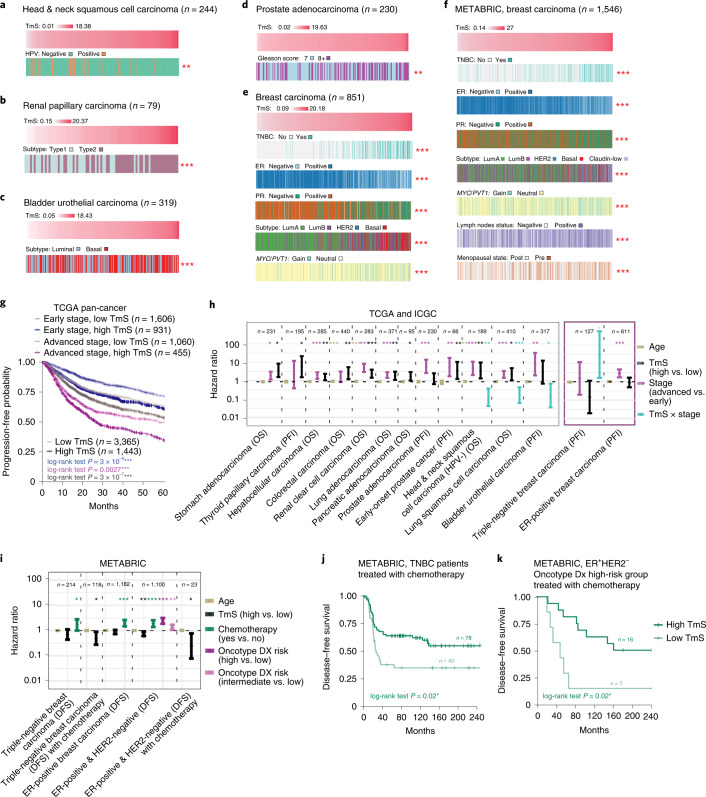
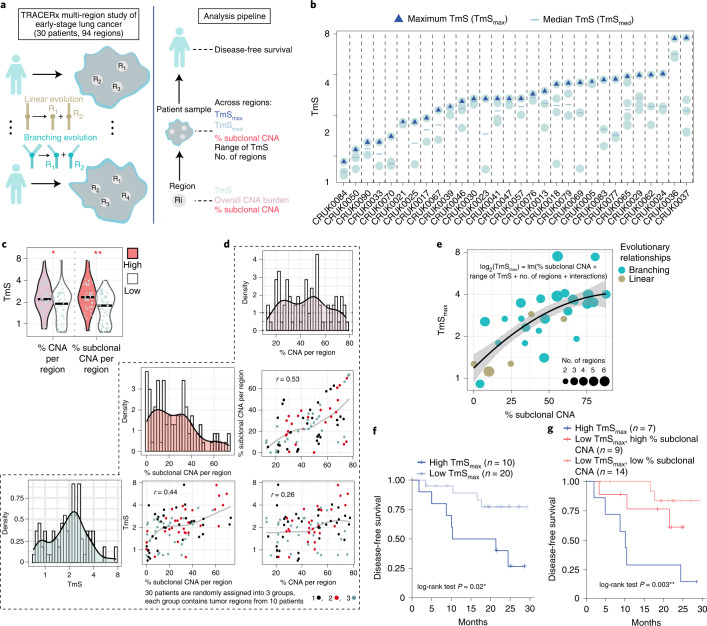
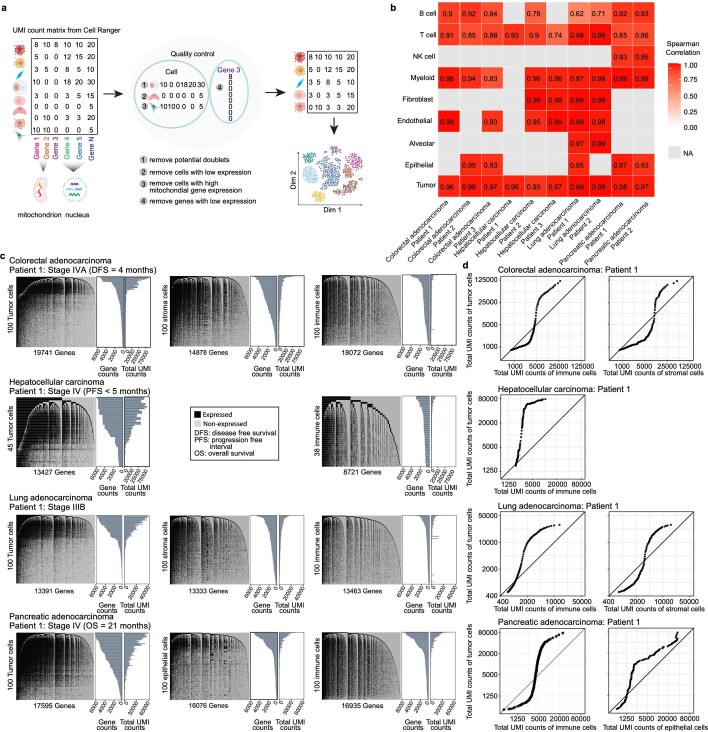
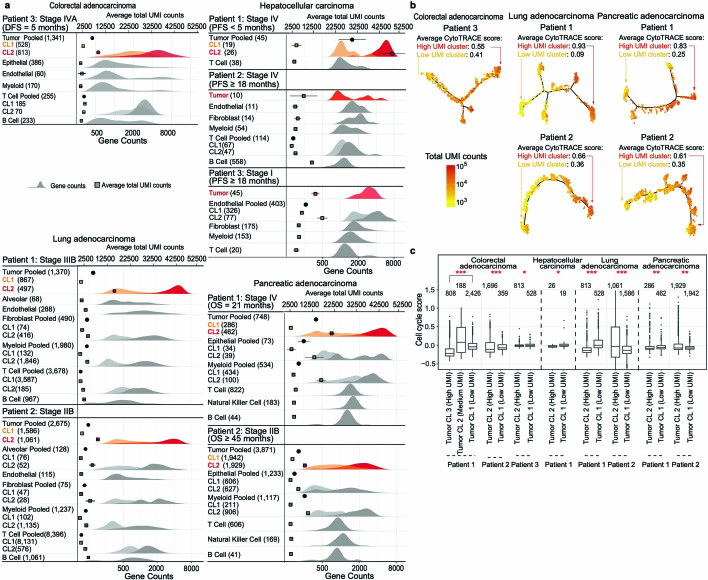
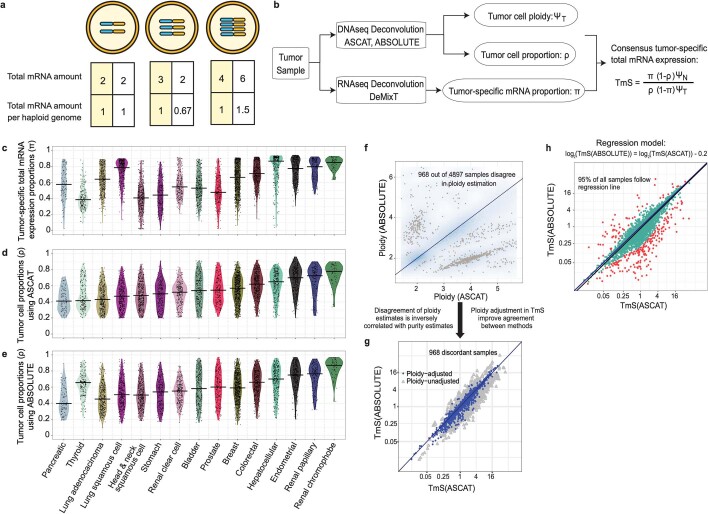
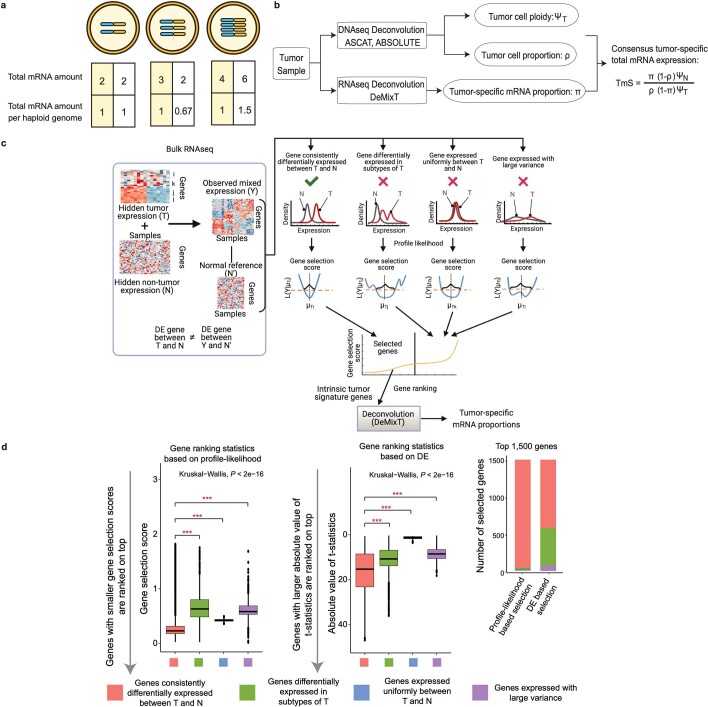
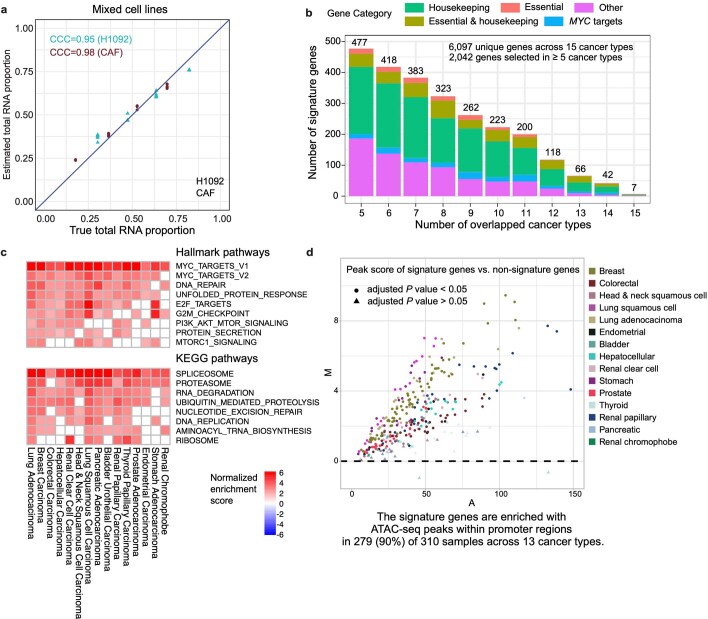
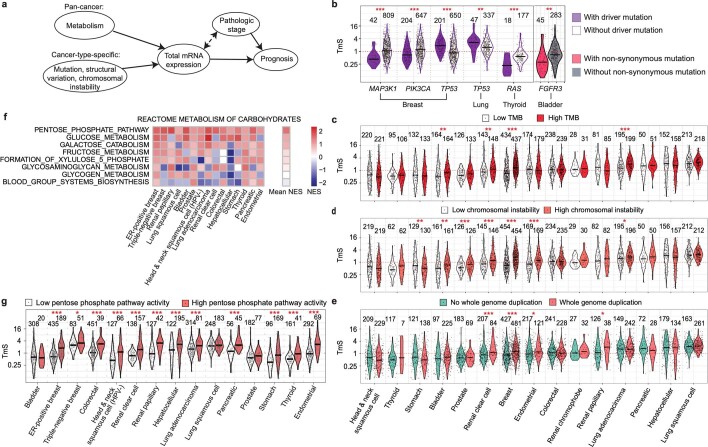
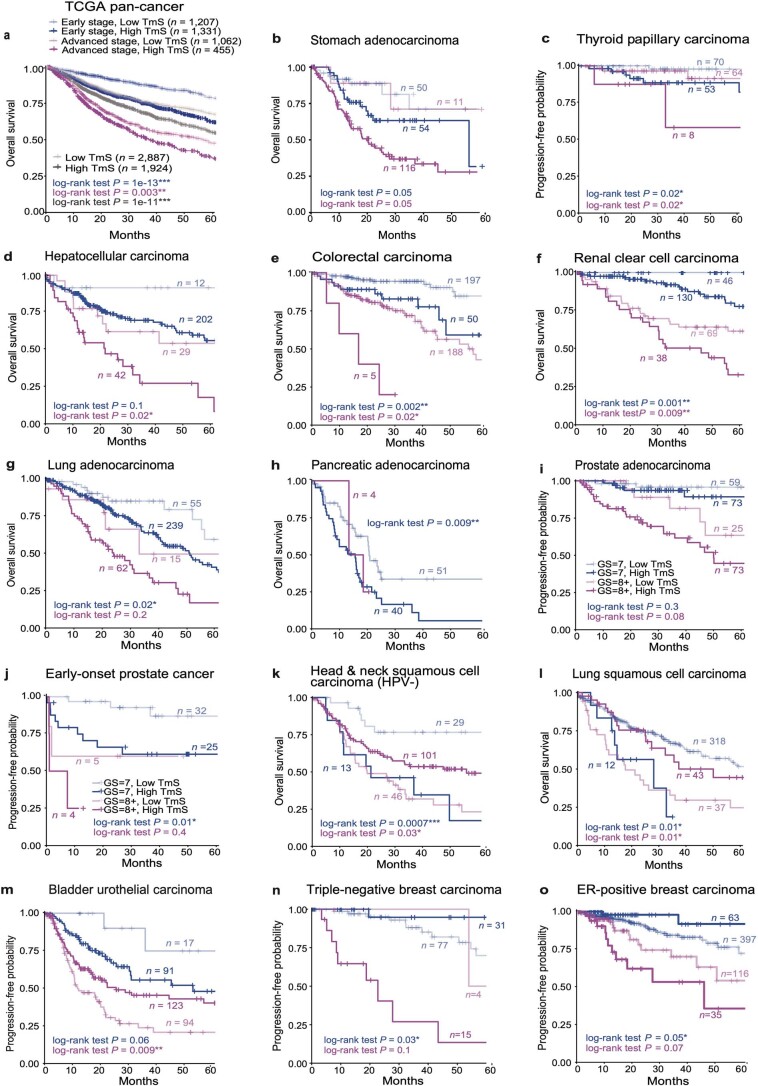
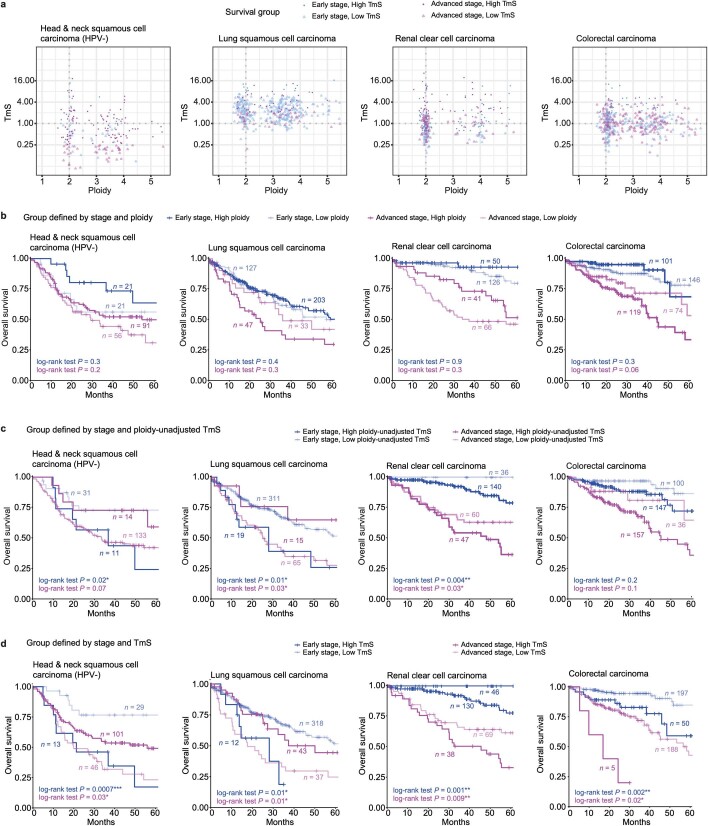
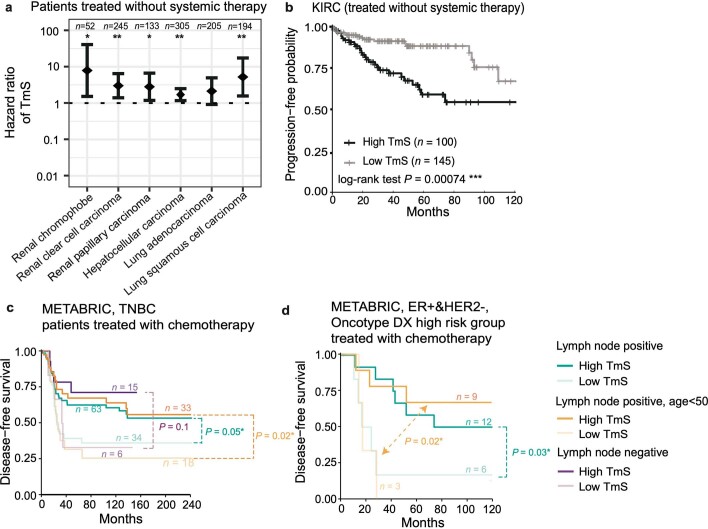
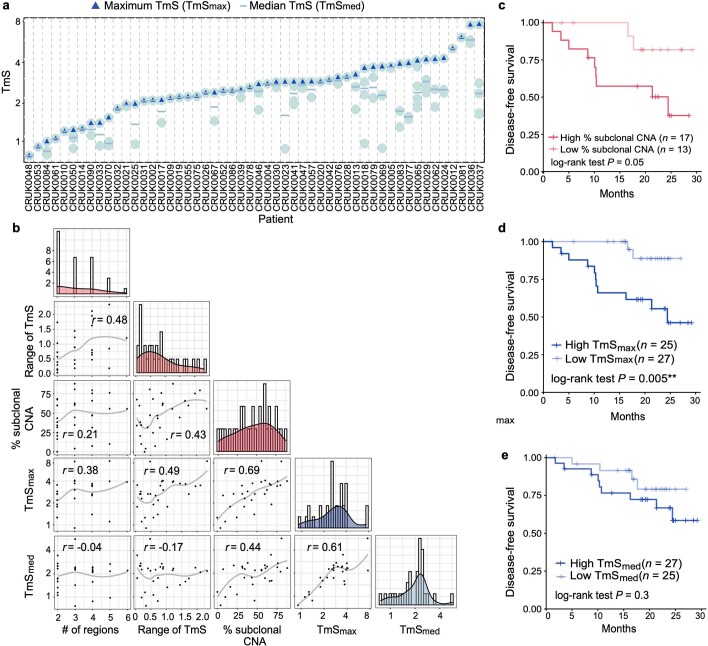
Single-cell RNA sequencing studies have suggested that total mRNA content correlates with tumor phenotypes. Technical and analytical challenges, however, have so far impeded at-scale pan-cancer examination of total mRNA content. Here we present a method to quantify tumor-specific total mRNA expression (TmS) from bulk sequencing data, taking into account tumor transcript proportion, purity and ploidy, which are estimated through transcriptomic/genomic deconvolution. We estimate and validate TmS in 6,590 patient tumors across 15 cancer types, identifying significant inter-tumor variability. Across cancers, high TmS is associated with increased risk of disease progression and death. TmS is influenced by cancer-specific patterns of gene alteration and intra-tumor genetic heterogeneity as well as by pan-cancer trends in metabolic dysregulation. Taken together, our results indicate that measuring cell-type-specific total mRNA expression in tumor cells predicts tumor phenotypes and clinical outcomes.
© 2022. The Author(s).
Conflict of interest statement
A.M. receives royalties for a pancreatic cancer biomarker test from Cosmos Wisdom Biotechnology. A.M. is also listed as an inventor on a patent that has been licensed by Johns Hopkins University to Thrive Earlier Detection. A.M. is a consultant for Freenome and Tezcat Biotechnology. J.Z. reports research funding from Merck and Johnson & Johnson and consultant fees from Bristol Myers Squibb (BMS), Johnson & Johnson, AstraZeneca, Geneplus, OrigMed and Innovent outside of the submitted work. P.M. has received honoraria for service on a Scientific Advisory Board for Mirati Therapeutics and BMS, non-branded educational programs supported by Exelixis and Pfizer and research funding for clinical trials from Takeda, BMS, Mirati Therapeutics and Gateway for Cancer Research. W.W. reports research funding from Curis, Inc. J.P.S. and W.W. report research funding from Celsius Therapeutics. J.P.S. is a paid consultant for Engine Biosciences. S.K. has ownership interest in MolecularMatch, Lutris and Iylon and is a consultant for Genentech, EMD Serono, Merck, Holy Stone, Novartis, Eli Lilly, Boehringer Ingelheim, Boston Biomedical, AstraZeneca/MedImmune, Bayer Health, Pierre Fabre, Redx Pharma, Ipsen, Daiichi Sankyo, Natera, HalioDx, Lutris, Jacobio, Pfizer, Repare Therapeutics, Inivata, GlaxoSmithKline, Jazz Pharmaceuticals, Iylon, Xilis, Abbvie, Amal Therapeutics, Gilead Sciences, Mirati Therapeutics, Flame Biosciences, Servier, Carina Biotechnology, Bicara Therapeutics, Endeavor BioMedicines, Numab Pharma and Johnson & Johnson/Janssen and receive research funding from Sanofi, Biocartis, Guardant Health, Array BioPharma, Genentech/Roche, EMD Serono, MedImmune, Novartis, Amgen, Eli Lilly and Daiichi Sankyo. P.A.F. reports research funding from MEI Pharma, Inc. P.H.B. owns stock in GeneTex. C.S. acknowledges grant support from AstraZeneca, Boehringer Ingelheim, BMS, Pfizer, Roche-Ventana, Invitae (previously Archer Dx—collaboration in minimal residual disease sequencing technologies) and Ono Pharmaceutical. C.S. is an AstraZeneca Advisory Board member and Chief Investigator for the AZ MeRmaiD 1 and 2 clinical trials and is also chief investigator of the NHS Galleri trial. C.S. has consulted for Amgen, AstraZeneca, Pfizer, Novartis, GlaxoSmithKline, Merck, BMS, Illumina, Genentech, Roche-Ventana, GRAIL, Medicxi, Metabomed, Bicycle Therapeutics, Roche Innovation Centre Shanghai and the Sarah Cannon Research Institute. C.S. had stock options in Apogen Biotechnologies and GRAIL until June 2021; currently has stock options in Epic Bioscience and Bicycle Therapeutics; and has stock options in and is a co-founder of Achilles Therapeutics. C.S. holds various patents relating to assay technology for cancer; US patents relating to detecting tumor mutations and methods for lung cancer detection; and both a European and a US patent related to identifying insertion/deletion mutation targets. All is outside the submitted work. The remaining authors declare no competing interests.
Figures
References
-
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U01 CA196403/CA/NCI NIH HHS/United States
- P30 CA016672/CA/NCI NIH HHS/United States
- U01 CA224044/CA/NCI NIH HHS/United States
- R01 CA268380/CA/NCI NIH HHS/United States
- DH_/Department of Health/United Kingdom
- FC001169/CRUK_/Cancer Research UK/United Kingdom
- C416/A21999/CRUK_/Cancer Research UK/United Kingdom
- MR/L016311/1/MRC_/Medical Research Council/United Kingdom
- R01 CA231465/CA/NCI NIH HHS/United States
- U01 CA256780/CA/NCI NIH HHS/United States
- R01 CA183793/CA/NCI NIH HHS/United States
- FC001202/MRC_/Medical Research Council/United Kingdom
- U01 CA200468/CA/NCI NIH HHS/United States
- FC001202/WT_/Wellcome Trust/United Kingdom
- R01 CA234629/CA/NCI NIH HHS/United States
- U01 CA214194/CA/NCI NIH HHS/United States
- FC001202/CRUK_/Cancer Research UK/United Kingdom
- P50 CA221707/CA/NCI NIH HHS/United States
- C11496/A17786/CRUK_/Cancer Research UK/United Kingdom
- L30 CA171000/CA/NCI NIH HHS/United States
- FC001169/WT_/Wellcome Trust/United Kingdom
- FC001169/MRC_/Medical Research Council/United Kingdom
- U24 CA248265/CA/NCI NIH HHS/United States
- K22 CA234406/CA/NCI NIH HHS/United States
- P30 CA016042/CA/NCI NIH HHS/United States
- C11496/A30025/CRUK_/Cancer Research UK/United Kingdom
- U24 CA224020/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
