

Niche-specific genome degradation and convergent evolution shaping Staphylococcus aureus adaptation during severe infections
- PMID: 35699423
- PMCID: PMC9270034
- DOI: 10.7554/eLife.77195
Niche-specific genome degradation and convergent evolution shaping Staphylococcus aureus adaptation during severe infections
Abstract
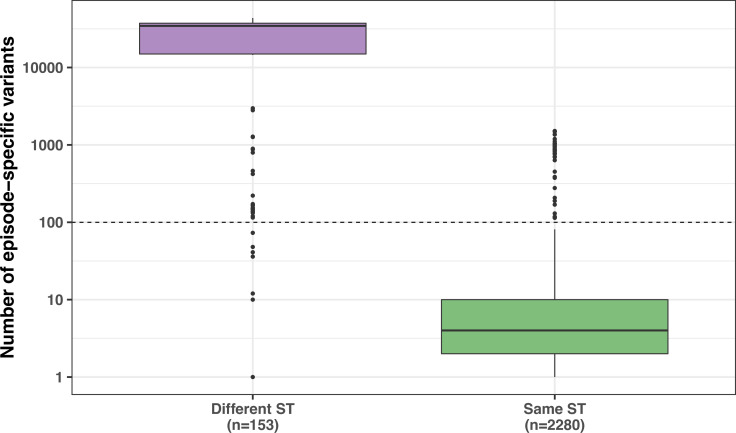
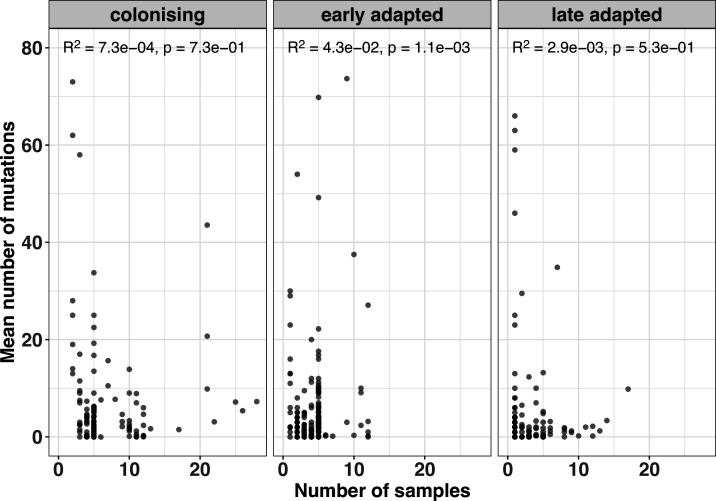
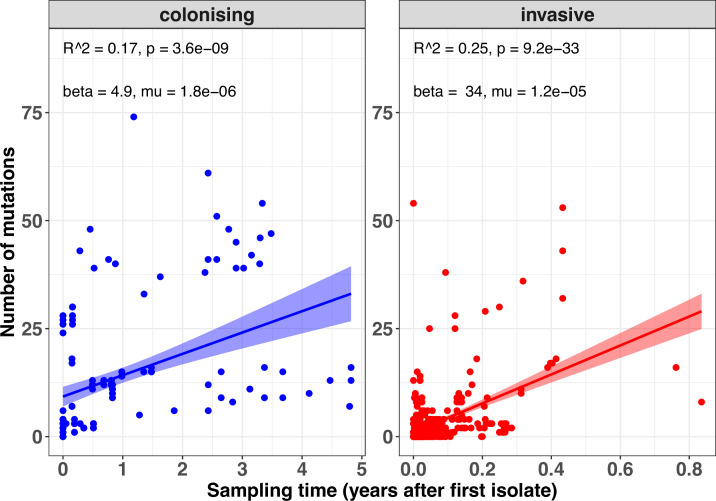
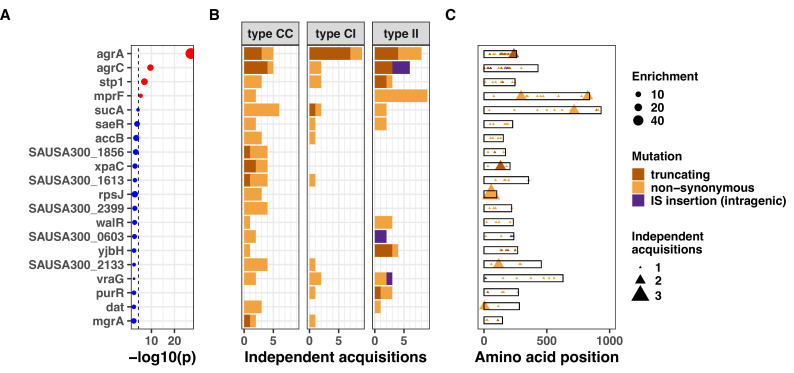
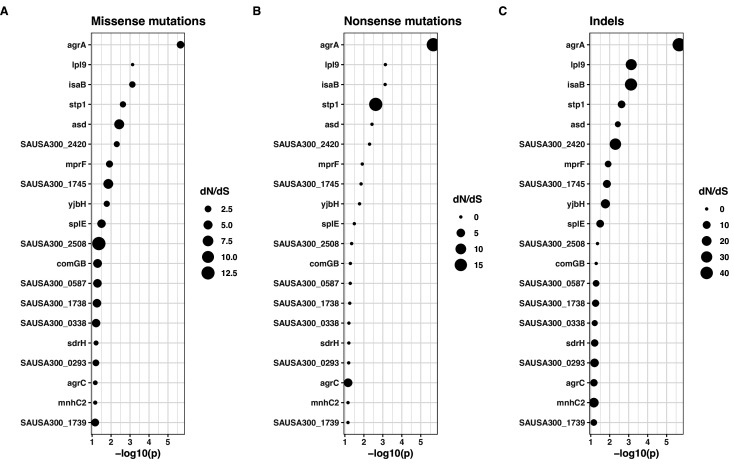
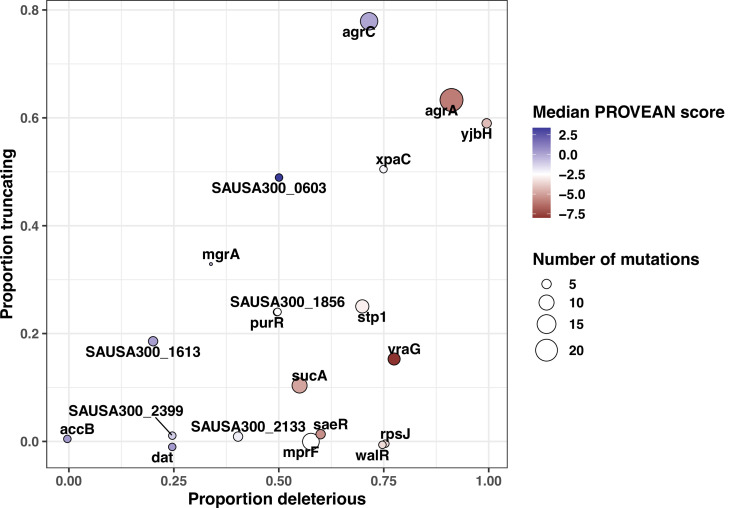
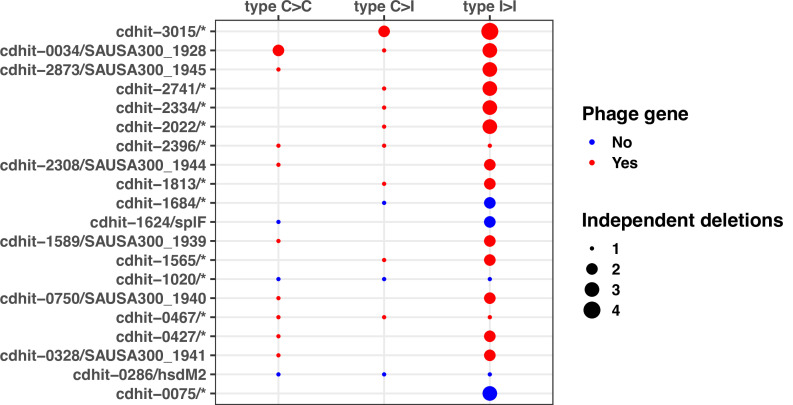
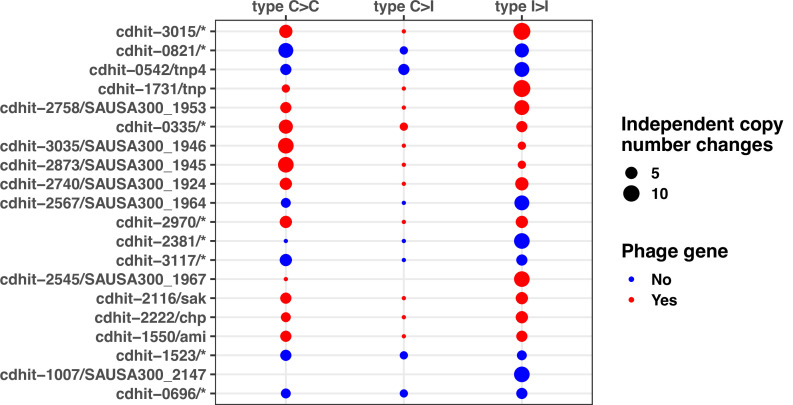
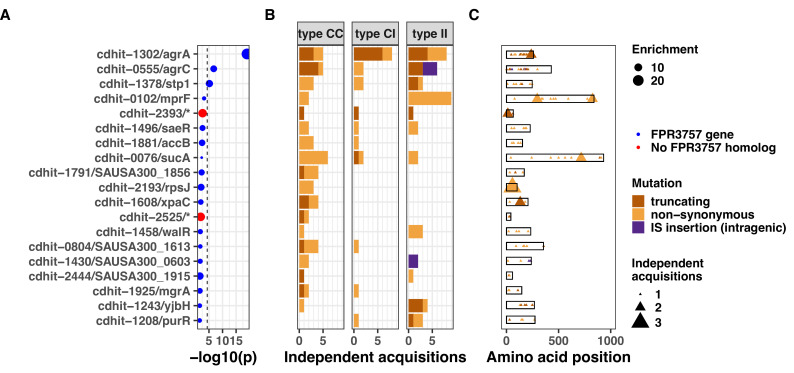
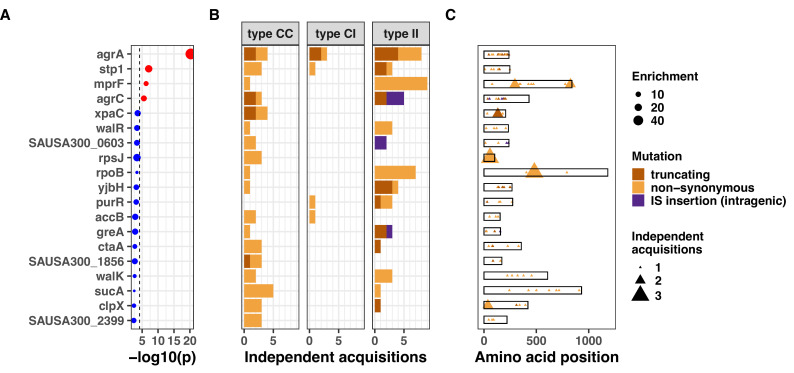
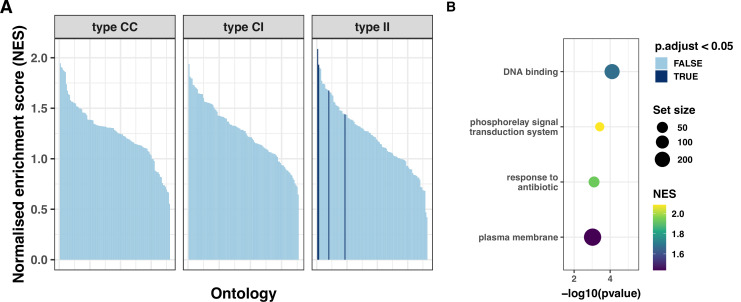
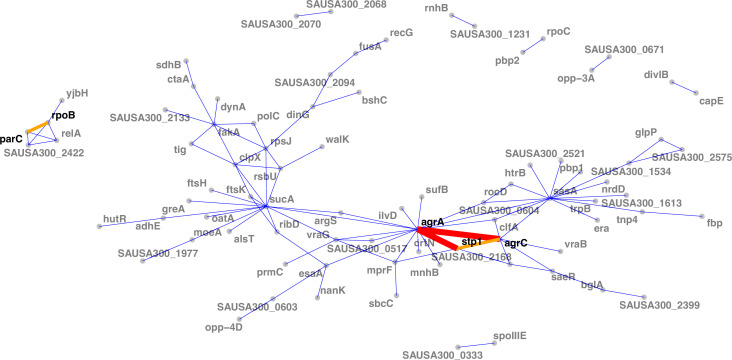
During severe infections, Staphylococcus aureus moves from its colonising sites to blood and tissues and is exposed to new selective pressures, thus, potentially driving adaptive evolution. Previous studies have shown the key role of the agr locus in S. aureus pathoadaptation; however, a more comprehensive characterisation of genetic signatures of bacterial adaptation may enable prediction of clinical outcomes and reveal new targets for treatment and prevention of these infections. Here, we measured adaptation using within-host evolution analysis of 2590 S. aureus genomes from 396 independent episodes of infection. By capturing a comprehensive repertoire of single nucleotide and structural genome variations, we found evidence of a distinctive evolutionary pattern within the infecting populations compared to colonising bacteria. These invasive strains had up to 20-fold enrichments for genome degradation signatures and displayed significantly convergent mutations in a distinctive set of genes, linked to antibiotic response and pathogenesis. In addition to agr-mediated adaptation, we identified non-canonical, genome-wide significant loci including sucA-sucB and stp1. The prevalence of adaptive changes increased with infection extent, emphasising the clinical significance of these signatures. These findings provide a high-resolution picture of the molecular changes when S. aureus transitions from colonisation to severe infection and may inform correlation of infection outcomes with adaptation signatures.
Keywords: Staphylococcus aureus; adaptation; genetics; genomics; infectious disease; microbiology; within-host evolution.
Plain language summary
The bacterium Staphylococcus aureus lives harmlessly on our skin and noses. However, occasionally, it gets into our blood and internal organs, such as our bones and joints, where it causes severe, long-lasting infections that are difficult to treat. Over time, S. aureus acquire characteristics that help them to adapt to different locations, such as transitioning from the nose to the blood, and avoid being killed by antibiotics. Previous studies have identified changes, or ‘mutations’, in genes that are likely to play an important role in this evolutionary process. One of these genes, called accessory gene regulator (or agr for short), has been shown to control the mechanisms S. aureus use to infect cells and disseminate in the body. However, it is unclear if there are changes in other genes that also help S. aureus adapt to life inside the human body. To help resolve this mystery, Giulieri et al. collected 2,500 samples of S. aureus from almost 400 people. This included bacteria harmlessly living on the skin or in the nose, as well as strains that caused an infection. Gene sequencing revealed a small number of genes, referred to as ‘adaptive genes’, that often acquire mutations during infection. Of these, agr was the most commonly altered. However, mutations in less well-known genes were also identified: some of these genes are related to resistance to antibiotics, while others are involved in chemical processes that help the bacteria to process nutrients. Most mutations were caused by random errors being introduced in to the bacteria’s genetic code which stopped genes from working. However, in some cases, genes were turned off by small fragments of DNA moving around and inserting themselves into different parts of the genome. This study highlights a group of genes that help S. aureus to thrive inside the body and cause severe and prolonged infections. If these results can be confirmed, it may help to guide which antibiotics are used to treat different infections. Furthermore, understanding which genes are important for infection could lead to new strategies for eliminating this dangerous bacterium.
© 2022, Giulieri et al.
Conflict of interest statement
SG, RG, SD, AH, DD, TS, JD, ST, BY, DW, TS, BH No competing interests declared
Figures










References
-
- Altman DR, Sullivan MJ, Chacko KI, Balasubramanian D, Pak TR, Sause WE, Kumar K, Sebra R, Deikus G, Attie O, Rose H, Lewis M, Fulmer Y, Bashir A, Kasarskis A, Schadt EE, Richardson AR, Torres VJ, Shopsin B, van Bakel H. Genome Plasticity of agr-Defective Staphylococcus aureus during Clinical Infection. Infection and Immunity. 2018;86:e00331-18. doi: 10.1128/IAI.00331-18. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Medical
