

ChIP-Hub provides an integrative platform for exploring plant regulome
- PMID: 35701419
- PMCID: PMC9197862
- DOI: 10.1038/s41467-022-30770-1
ChIP-Hub provides an integrative platform for exploring plant regulome
Abstract
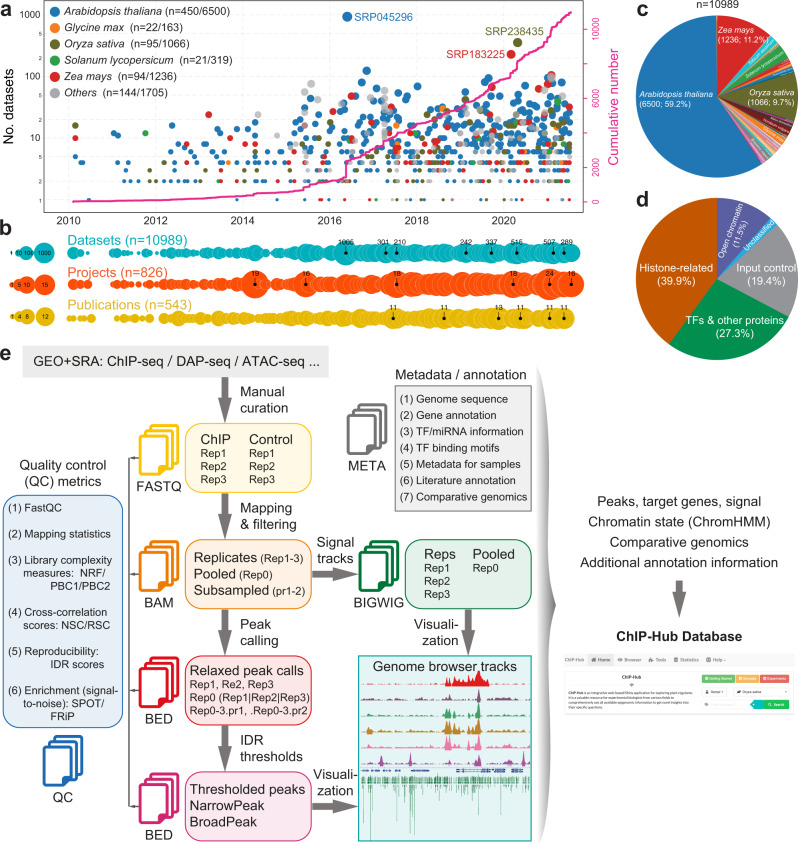
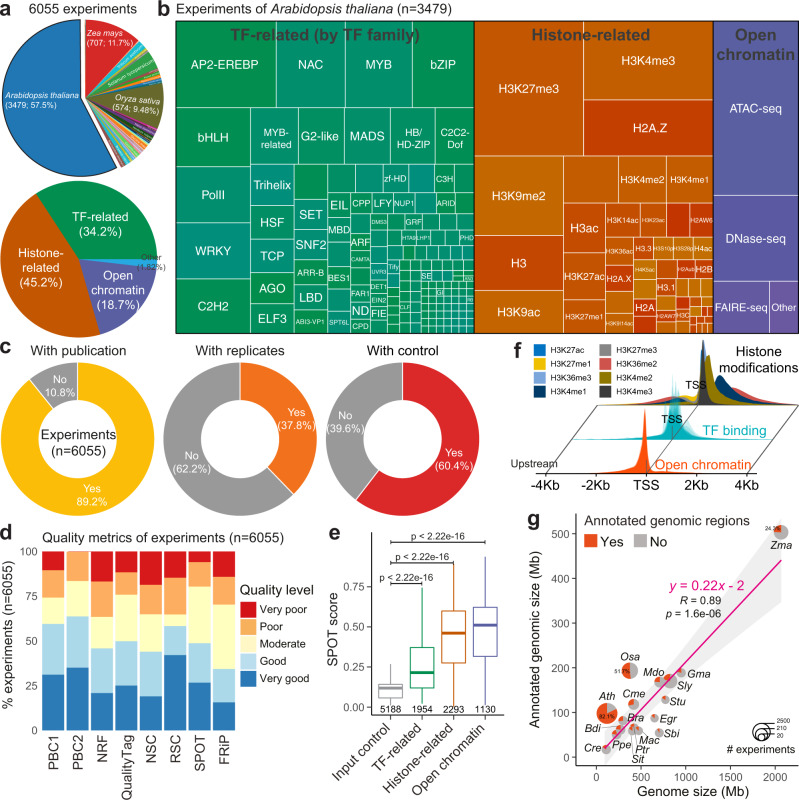
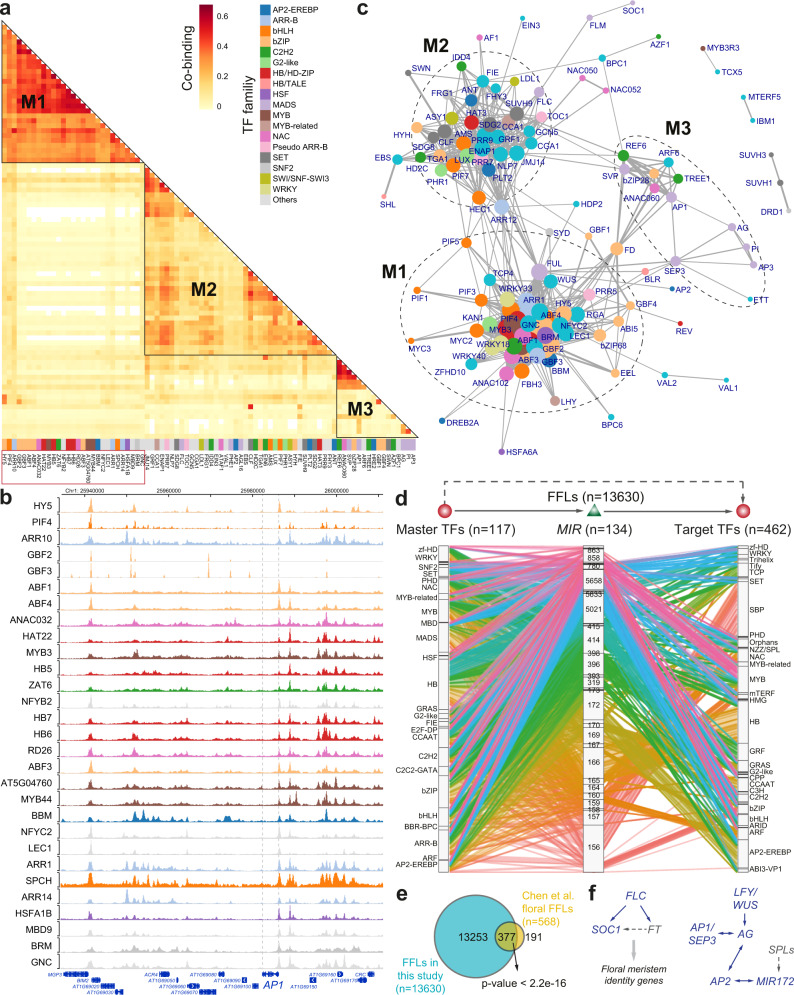
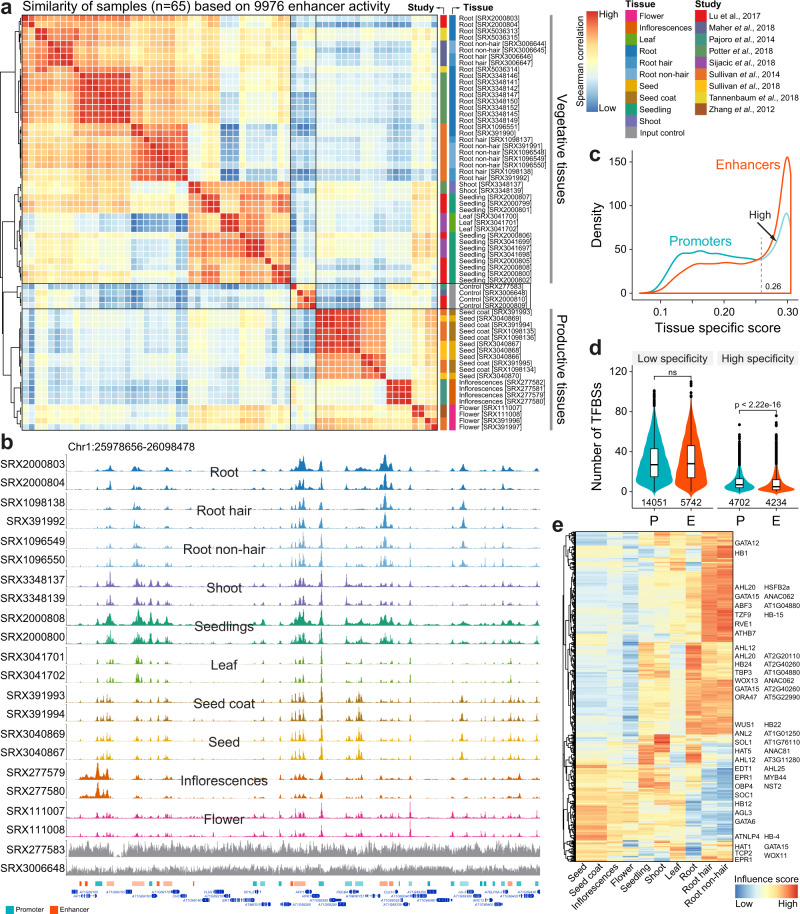
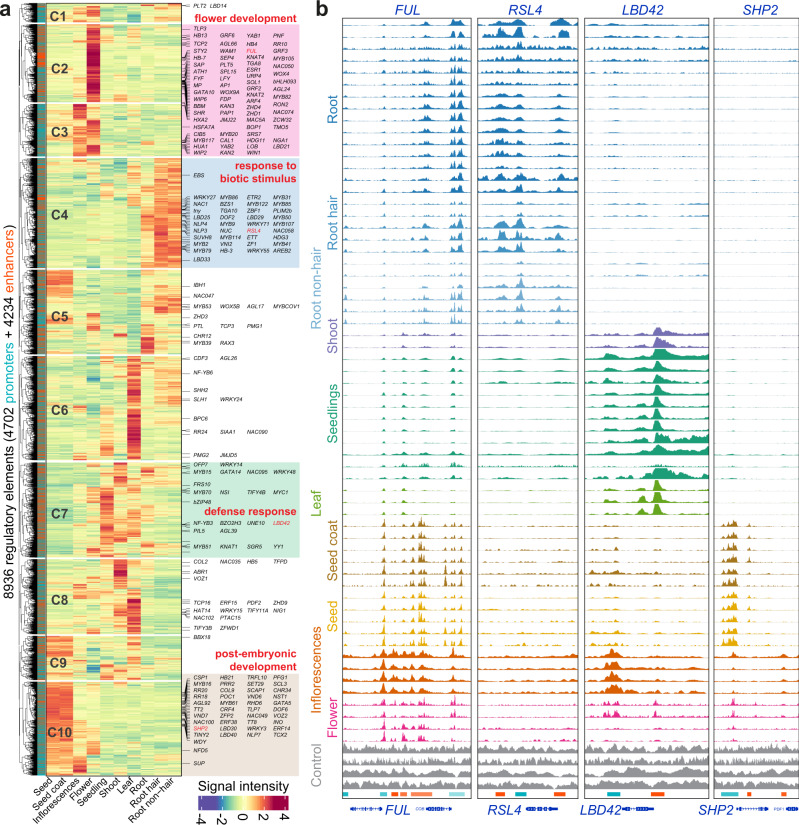
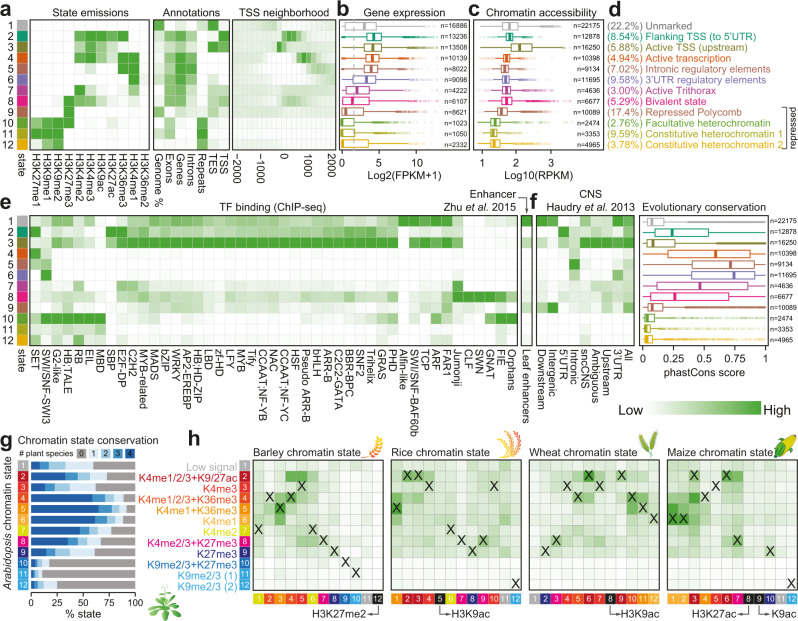
Plant genomes encode a complex and evolutionary diverse regulatory grammar that forms the basis for most life on earth. A wealth of regulome and epigenome data have been generated in various plant species, but no common, standardized resource is available so far for biologists. Here, we present ChIP-Hub, an integrative web-based platform in the ENCODE standards that bundles >10,000 publicly available datasets reanalyzed from >40 plant species, allowing visualization and meta-analysis. We manually curate the datasets through assessing ~540 original publications and comprehensively evaluate their data quality. As a proof of concept, we extensively survey the co-association of different regulators and construct a hierarchical regulatory network under a broad developmental context. Furthermore, we show how our annotation allows to investigate the dynamic activity of tissue-specific regulatory elements (promoters and enhancers) and their underlying sequence grammar. Finally, we analyze the function and conservation of tissue-specific promoters, enhancers and chromatin states using comparative genomics approaches. Taken together, the ChIP-Hub platform and the analysis results provide rich resources for deep exploration of plant ENCODE. ChIP-Hub is available at https://biobigdata.nju.edu.cn/ChIPHub/ .
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Johnson, D. S., Mortazavi, A., Myers, R. M. & Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science (80-.). 10.1126/science.1141319 (2007). - PubMed
-
- Barski, A. et al. High-resolution profiling of histone methylations in the human genome. Cell10.1016/j.cell.2007.05.009 (2007). - PubMed
-
- Robertson, G. et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods10.1038/nmeth1068 (2007). - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
