

Noncanonical Sonic Hedgehog signaling amplifies platelet reactivity and thrombogenicity
- PMID: 35704688
- PMCID: PMC9631642
- DOI: 10.1182/bloodadvances.2021006560
Noncanonical Sonic Hedgehog signaling amplifies platelet reactivity and thrombogenicity
Abstract
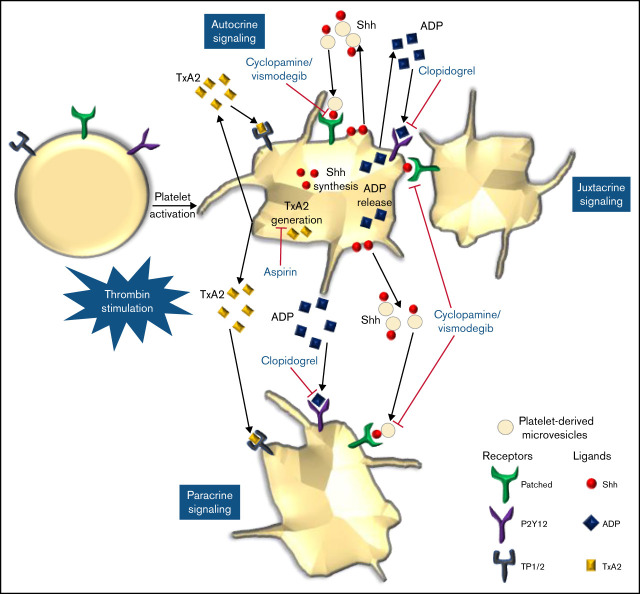
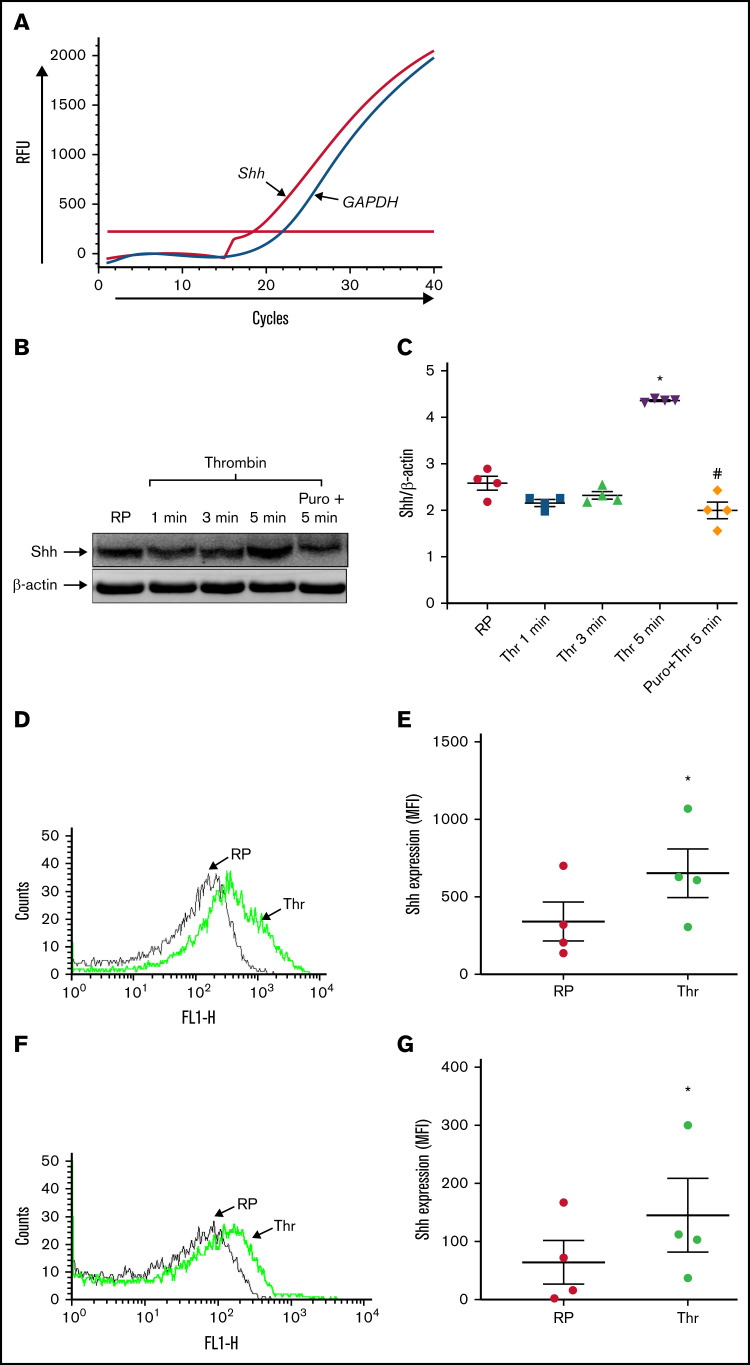
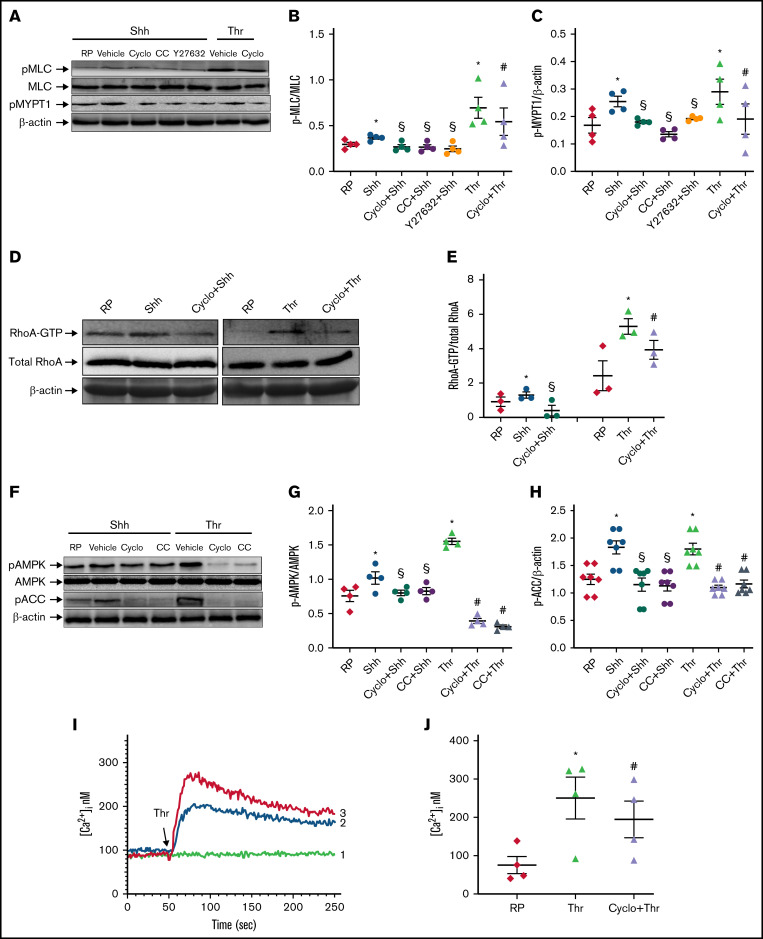
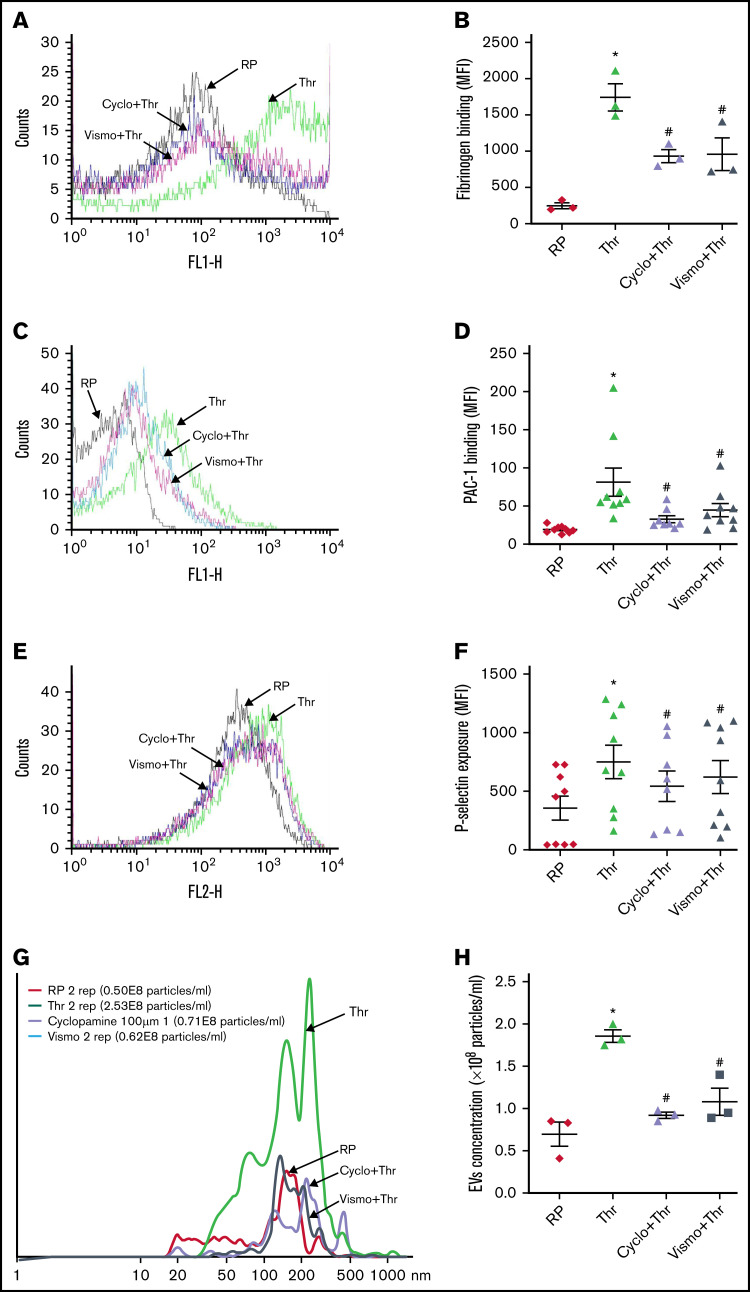
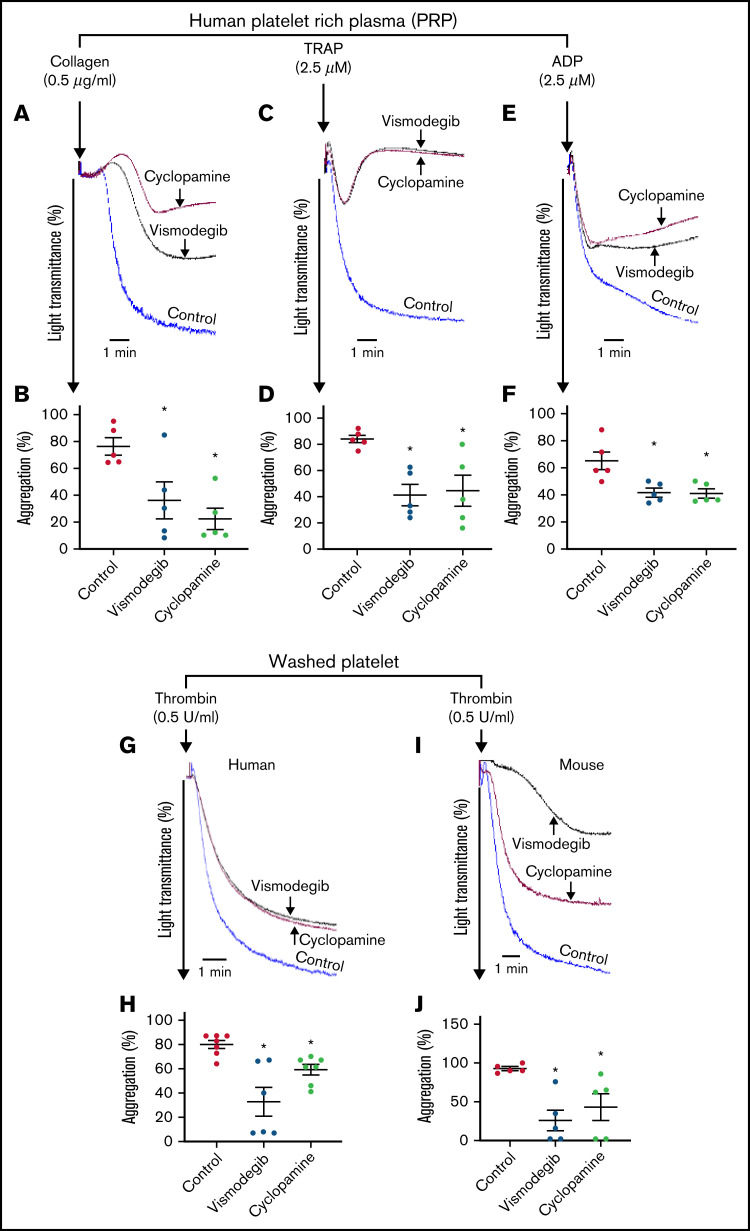
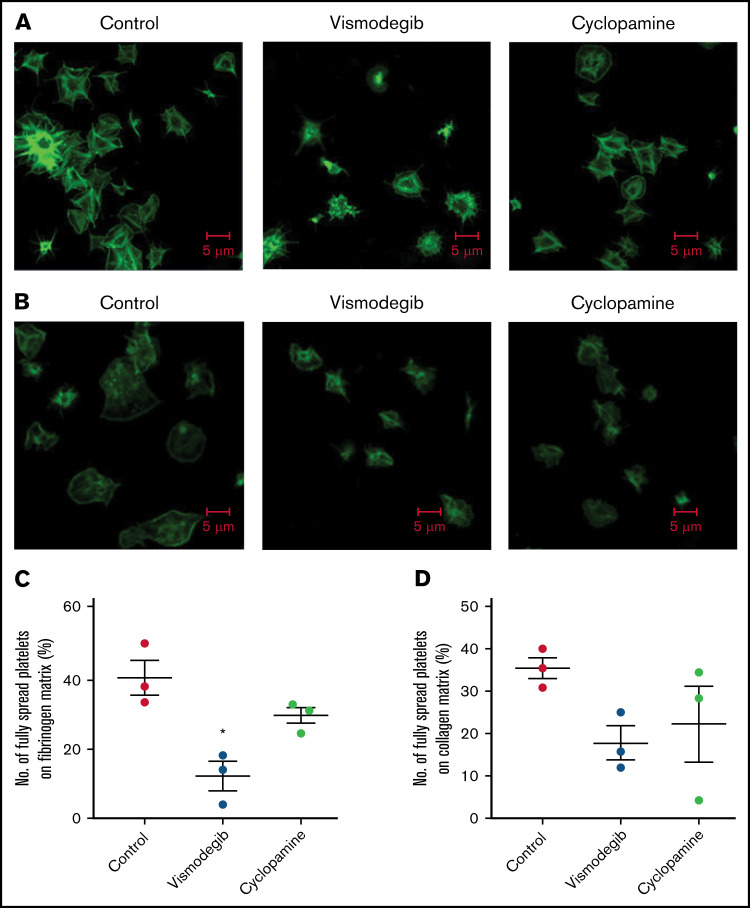
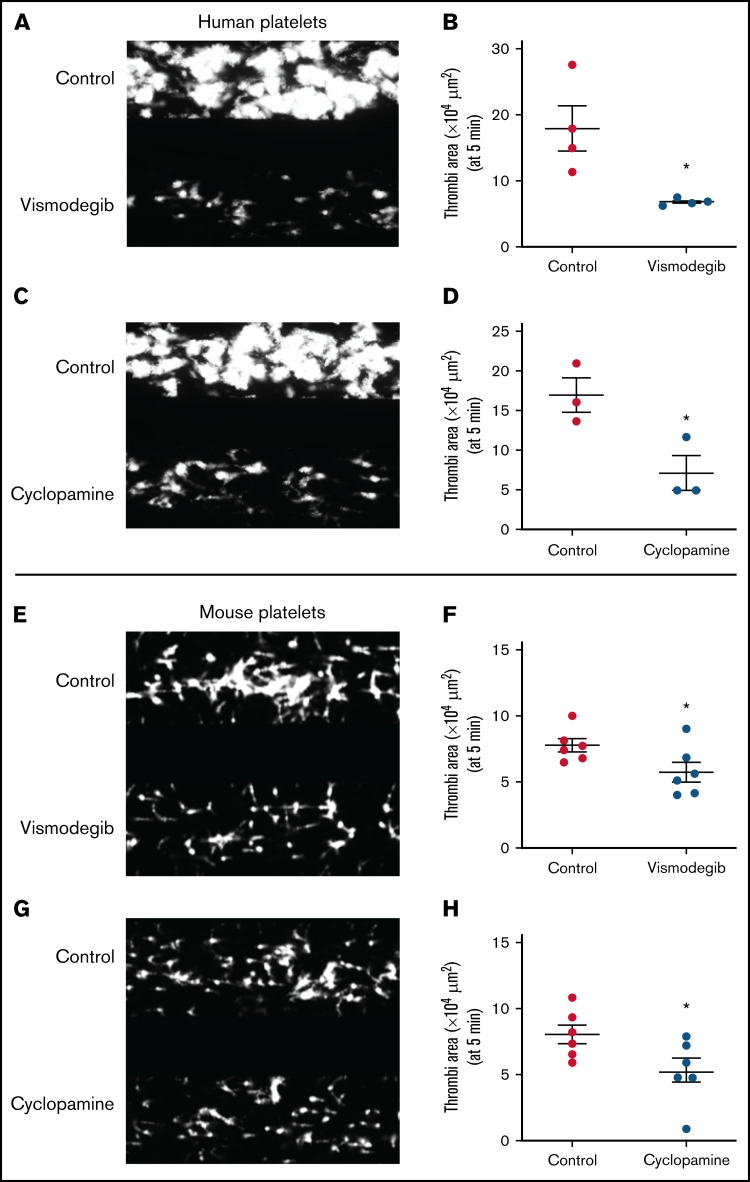
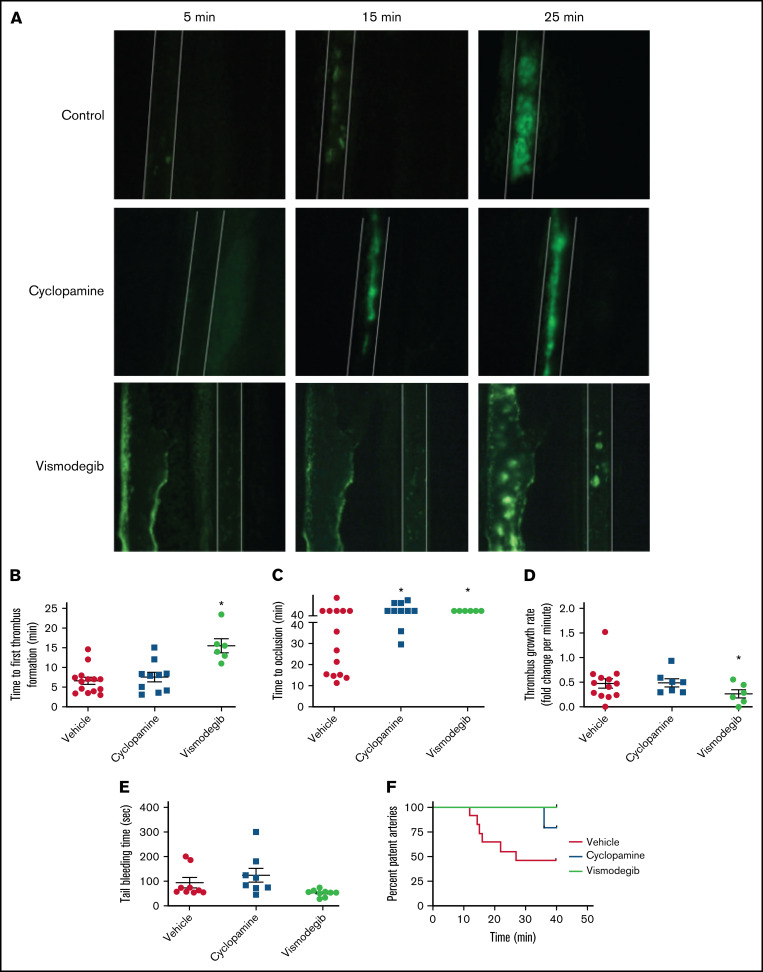
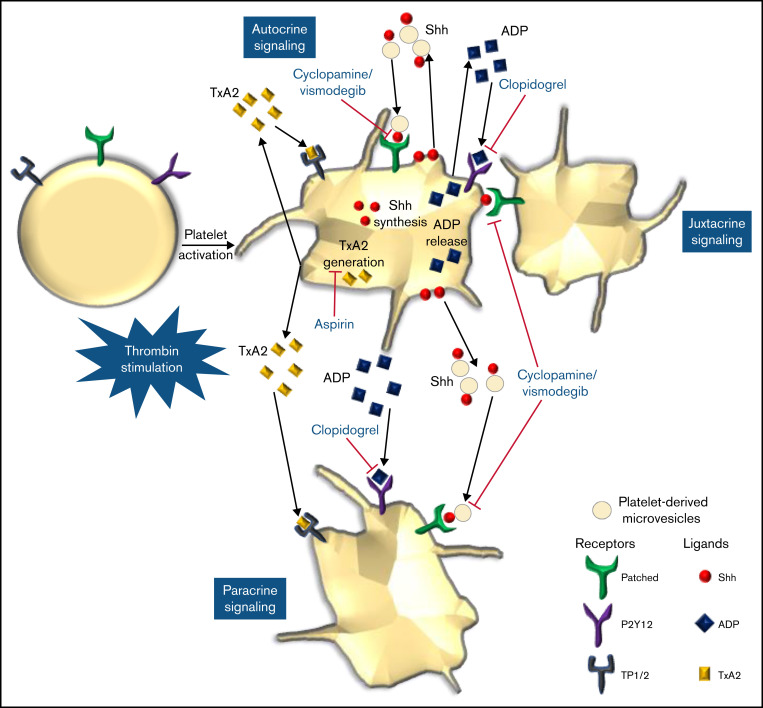
Sonic Hedgehog (Shh) is a morphogen in vertebrate embryos that is also associated with organ homeostasis in adults. We report here that human platelets, though enucleate, synthesize Shh from preexisting mRNAs upon agonist stimulation, and mobilize it for surface expression and release on extracellular vesicles, thus alluding to its putative role in platelet activation. Shh, in turn, induced a wave of noncanonical signaling in platelets leading to activation of small GTPase Ras homolog family member A and phosphorylation of myosin light chain in activated protein kinase-dependent manner. Remarkably, agonist-induced thrombogenic responses in platelets, which include platelet aggregation, granule secretion, and spreading on immobilized fibrinogen, were significantly attenuated by inhibition of Hedgehog signaling, thus, implicating inputs from Shh in potentiation of agonist-mediated platelet activation. In consistence, inhibition of the Shh pathway significantly impaired arterial thrombosis in mice. Taken together, the above observations strongly support a feed-forward loop of platelet stimulation triggered locally by Shh, similar to ADP and thromboxane A2, that contributes significantly to the stability of occlusive arterial thrombus and that can be investigated as a potential therapeutic target in thrombotic disorders.
© 2022 by The American Society of Hematology. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Figures
References
-
- Spinelli SL, Maggirwar SB, Blumberg N, Phipps RP. Nuclear emancipation: a platelet tour de force. Sci Signal. 2010;3(144):pe37. - PubMed
-
- Kumari S, Dash D. Regulation of β-catenin stabilization in human platelets [published correction appears in Biochimie. 2019;162:239]. Biochimie. 2013;95(6):1252-1257. - PubMed
-
- Fodde R, Brabletz T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol. 2007;19(2):150-158. - PubMed
-
- Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression [published correction appears in Nat Genet. 2001;29(3):351]. Nat Genet. 2001;29(2):117-129. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
