

Primary and secondary functions of HLA-E are determined by stability and conformation of the peptide-bound complexes
- PMID: 35705051
- PMCID: PMC9380258
- DOI: 10.1016/j.celrep.2022.110959
Primary and secondary functions of HLA-E are determined by stability and conformation of the peptide-bound complexes
Abstract
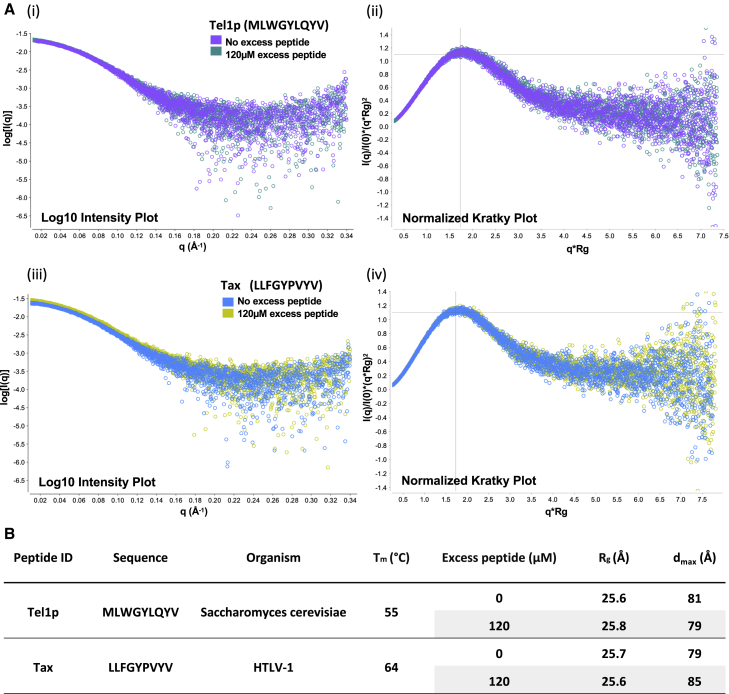
MHC-E regulates NK cells by displaying MHC class Ia signal peptides (VL9) to NKG2A:CD94 receptors. MHC-E can also present sequence-diverse, lower-affinity, pathogen-derived peptides to T cell receptors (TCRs) on CD8+ T cells. To understand these affinity differences, human MHC-E (HLA-E)-VL9 versus pathogen-derived peptide structures are compared. Small-angle X-ray scatter (SAXS) measures biophysical parameters in solution, allowing comparison with crystal structures. For HLA-E-VL9, there is concordance between SAXS and crystal parameters. In contrast, HLA-E-bound pathogen-derived peptides produce larger SAXS dimensions that reduce to their crystallographic dimensions only when excess peptide is supplied. Further crystallographic analysis demonstrates three amino acids, exclusive to MHC-E, that not only position VL9 close to the α2 helix, but also allow non-VL9 peptide binding with re-configuration of a key TCR-interacting α2 region. Thus, non-VL9-bound peptides introduce an alternative peptide-binding motif and surface recognition landscape, providing a likely basis for VL9- and non-VL9-HLA-E immune discrimination.
Keywords: CD8 T cells; CP: Immunology; HLA-E; MHC Ia; MHC-E; NK cells; NKG2A; SAXS; T cell receptor; VL9; X-ray crystallography; small-angle X-ray scatter.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures






References
-
- Afonine P.V., Grosse-Kunstleve R.W., Echols N., Headd J.J., Moriarty N.W., Mustyakimov M., Terwilliger T.C., Urzhumtsev A., Zwart P.H., Adams P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta. Crystallogr. D. Biol. Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. - DOI - PMC - PubMed
-
- Anjanappa R., Garcia-Alai M., Kopicki J.D., Lockhauserbäumer J., Aboelmagd M., Hinrichs J., Nemtanu I.M., Uetrecht C., Zacharias M., Springer S., Meijers R. Structures of peptide-free and partially loaded MHC class I molecules reveal mechanisms of peptide selection. Nat. Commun. 2020;11:1314. doi: 10.1038/s41467-020-14862-4. - DOI - PMC - PubMed
-
- Birkinshaw R.W., Pellicci D.G., Cheng T.Y., Keller A.N., Sandoval-Romero M., Gras S., de Jong A., Uldrich A.P., Moody D.B., Godfrey D.I., Rossjohn J. αβ T cell antigen receptor recognition of CD1a presenting self lipid ligands. Nat. Immunol. 2015;16:258–266. doi: 10.1038/ni.3098. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
