

Signatures of copy number alterations in human cancer
- PMID: 35705804
- PMCID: PMC9242861
- DOI: 10.1038/s41586-022-04738-6
Signatures of copy number alterations in human cancer
Abstract
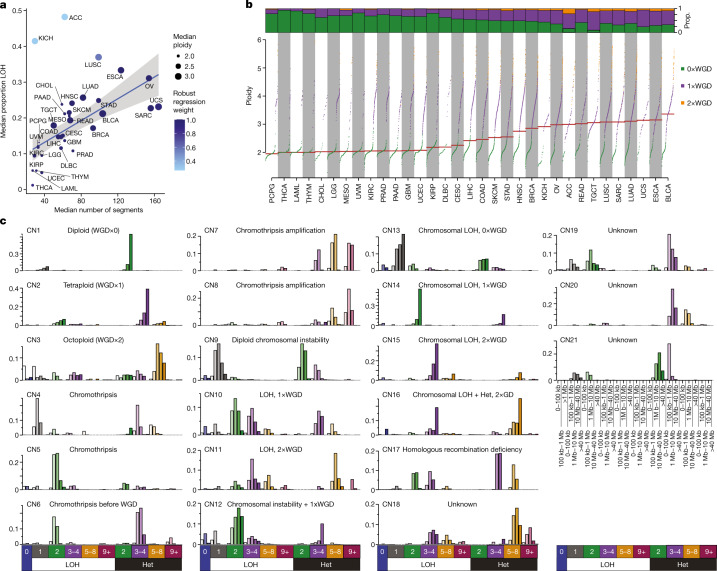
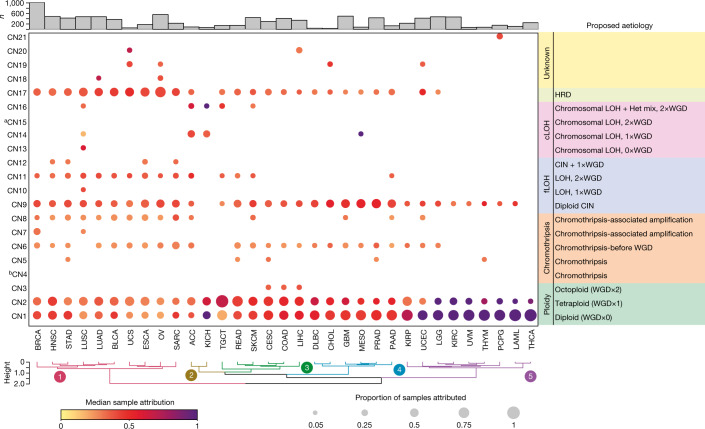
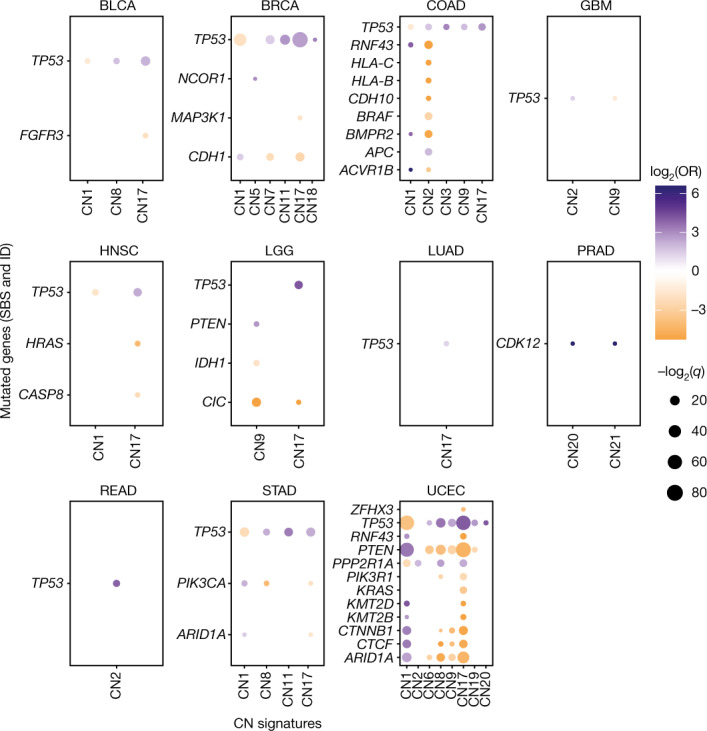
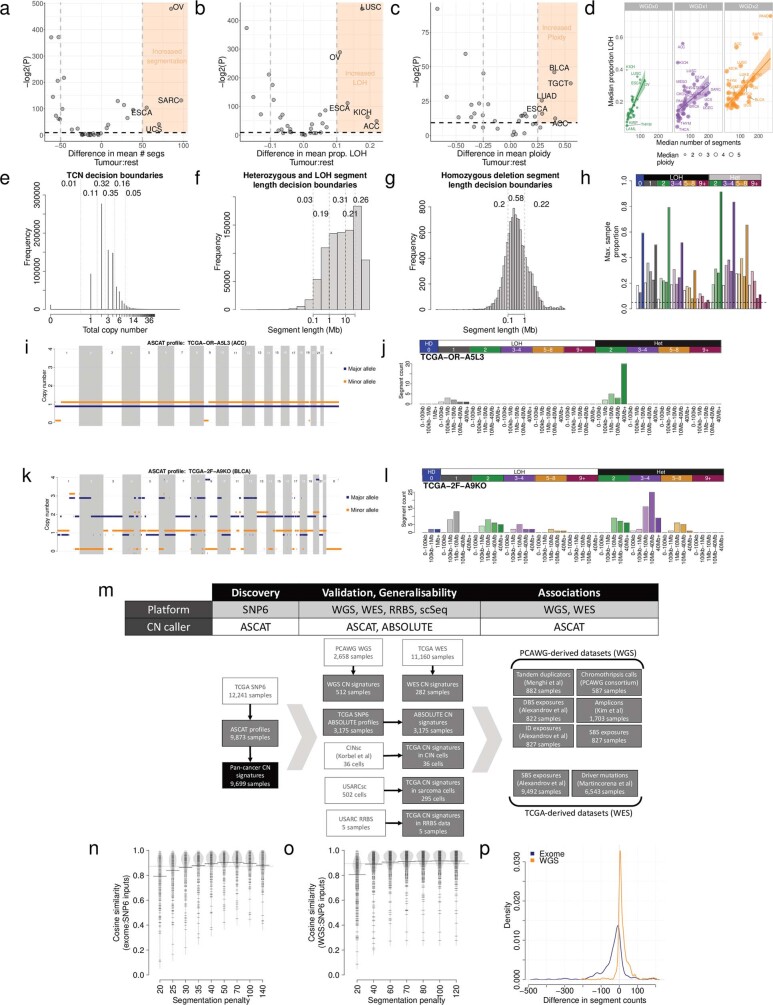
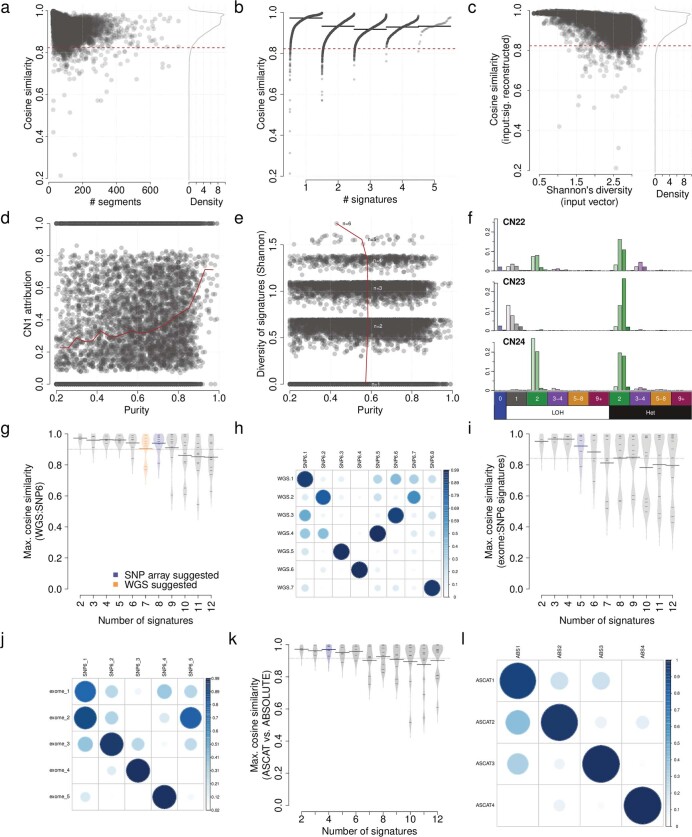
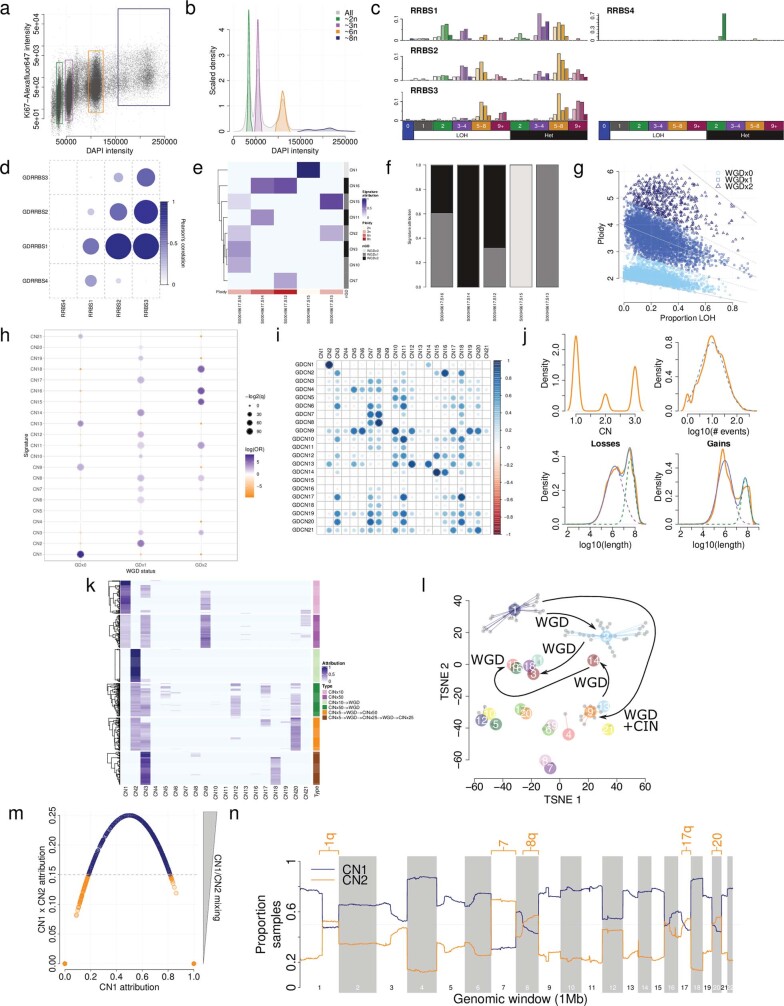
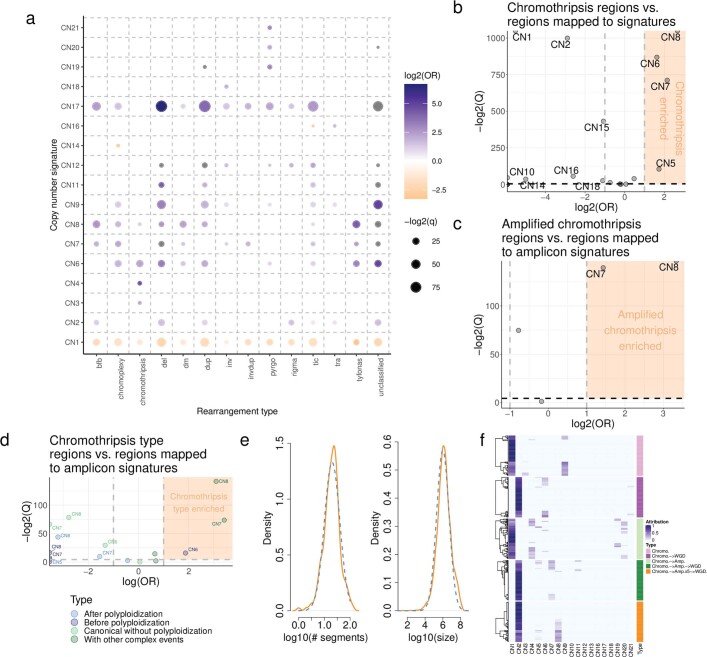
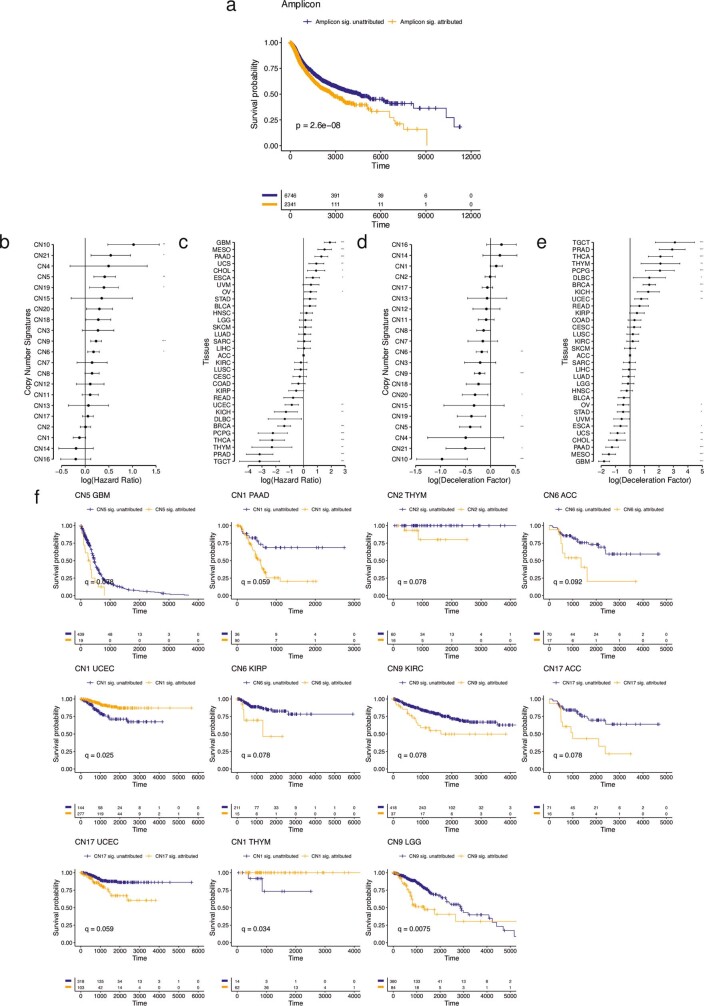
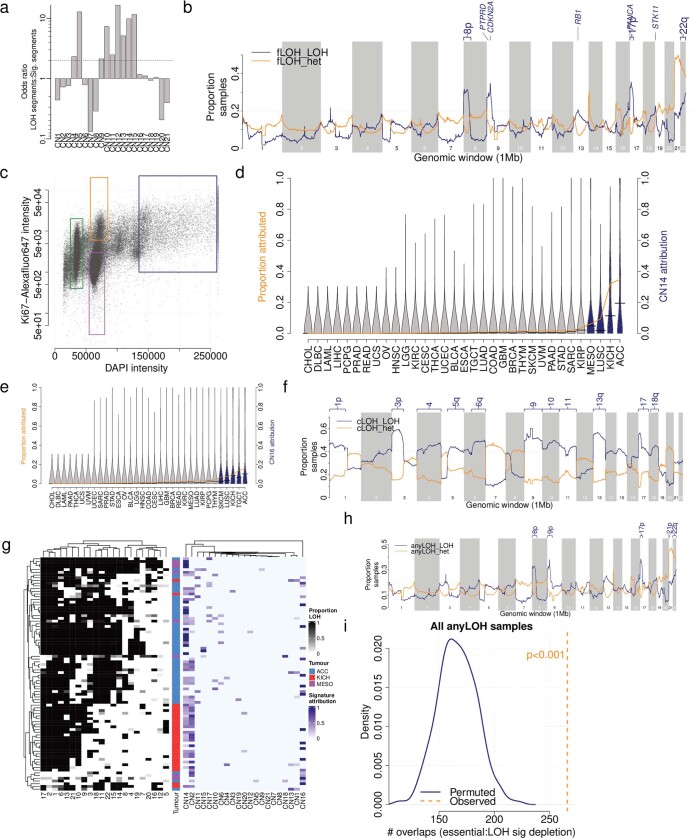
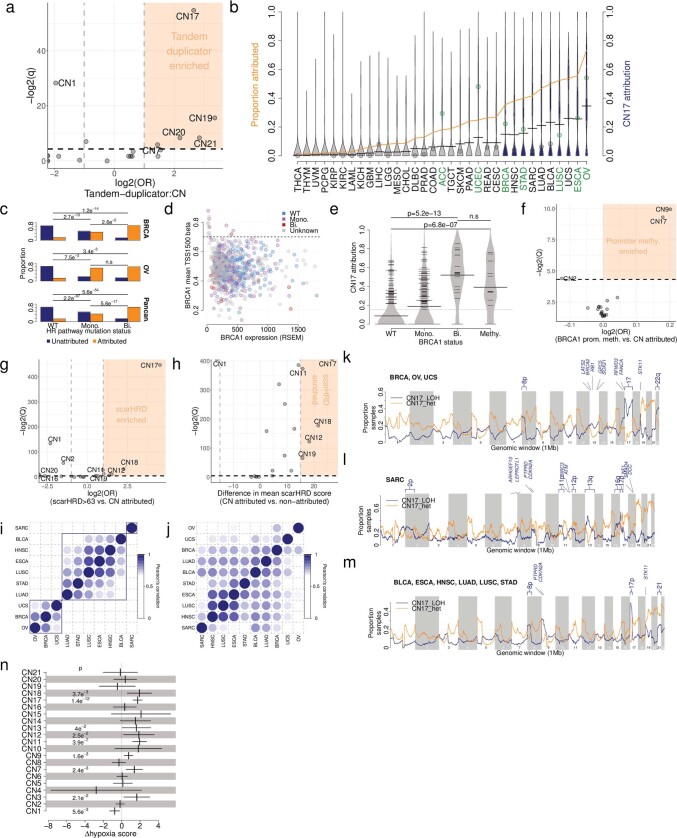
Gains and losses of DNA are prevalent in cancer and emerge as a consequence of inter-related processes of replication stress, mitotic errors, spindle multipolarity and breakage-fusion-bridge cycles, among others, which may lead to chromosomal instability and aneuploidy1,2. These copy number alterations contribute to cancer initiation, progression and therapeutic resistance3-5. Here we present a conceptual framework to examine the patterns of copy number alterations in human cancer that is widely applicable to diverse data types, including whole-genome sequencing, whole-exome sequencing, reduced representation bisulfite sequencing, single-cell DNA sequencing and SNP6 microarray data. Deploying this framework to 9,873 cancers representing 33 human cancer types from The Cancer Genome Atlas6 revealed a set of 21 copy number signatures that explain the copy number patterns of 97% of samples. Seventeen copy number signatures were attributed to biological phenomena of whole-genome doubling, aneuploidy, loss of heterozygosity, homologous recombination deficiency, chromothripsis and haploidization. The aetiologies of four copy number signatures remain unexplained. Some cancer types harbour amplicon signatures associated with extrachromosomal DNA, disease-specific survival and proto-oncogene gains such as MDM2. In contrast to base-scale mutational signatures, no copy number signature was associated with many known exogenous cancer risk factors. Our results synthesize the global landscape of copy number alterations in human cancer by revealing a diversity of mutational processes that give rise to these alterations.
© 2022. The Author(s).
Conflict of interest statement
L.B.A. is an inventor on US Patent 10,776,718 for source identification by NMF. All other authors declare no competing interests.
Figures


Comment in
-
Copy-number classifiers for cancer.Nat Rev Genet. 2022 Aug;23(8):457. doi: 10.1038/s41576-022-00516-2. Nat Rev Genet. 2022. PMID: 35764797 No abstract available.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
