

MERTK activation drives osimertinib resistance in EGFR-mutant non-small cell lung cancer
- PMID: 35708914
- PMCID: PMC9337831
- DOI: 10.1172/JCI150517
MERTK activation drives osimertinib resistance in EGFR-mutant non-small cell lung cancer
Abstract
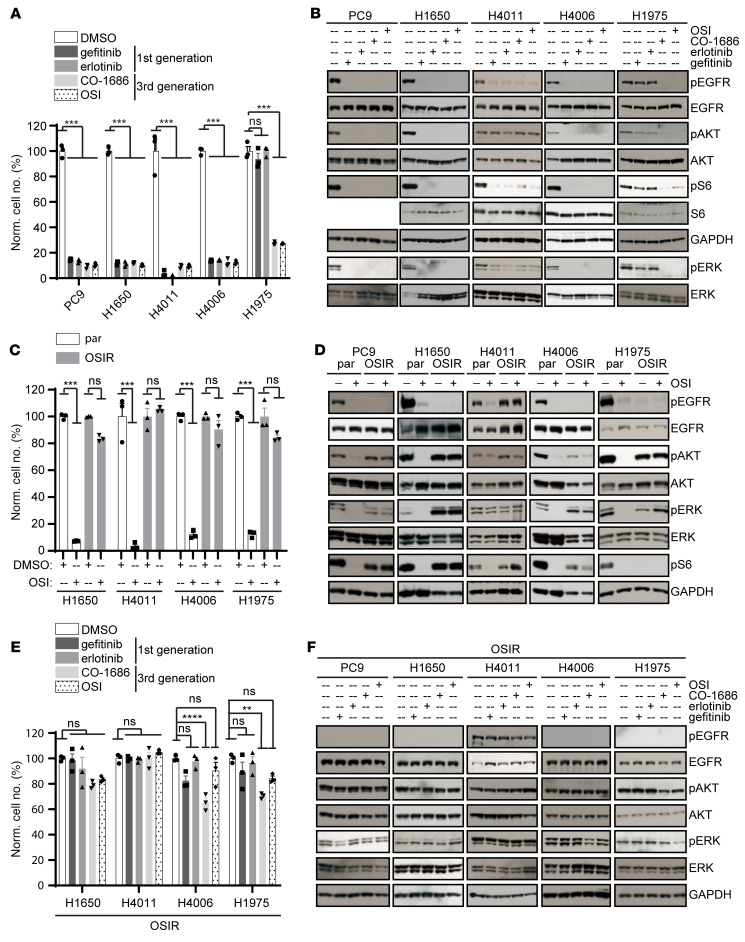
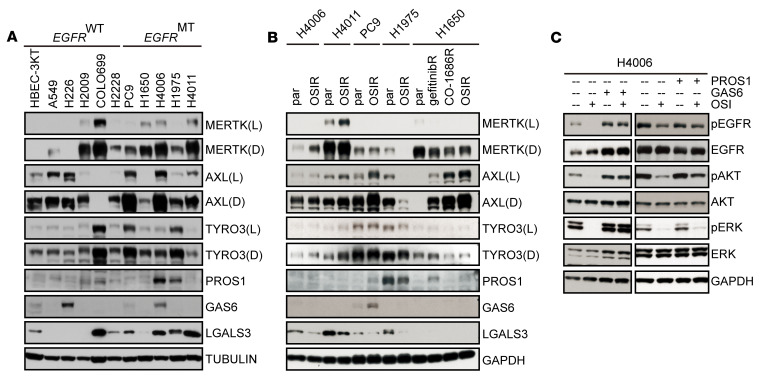
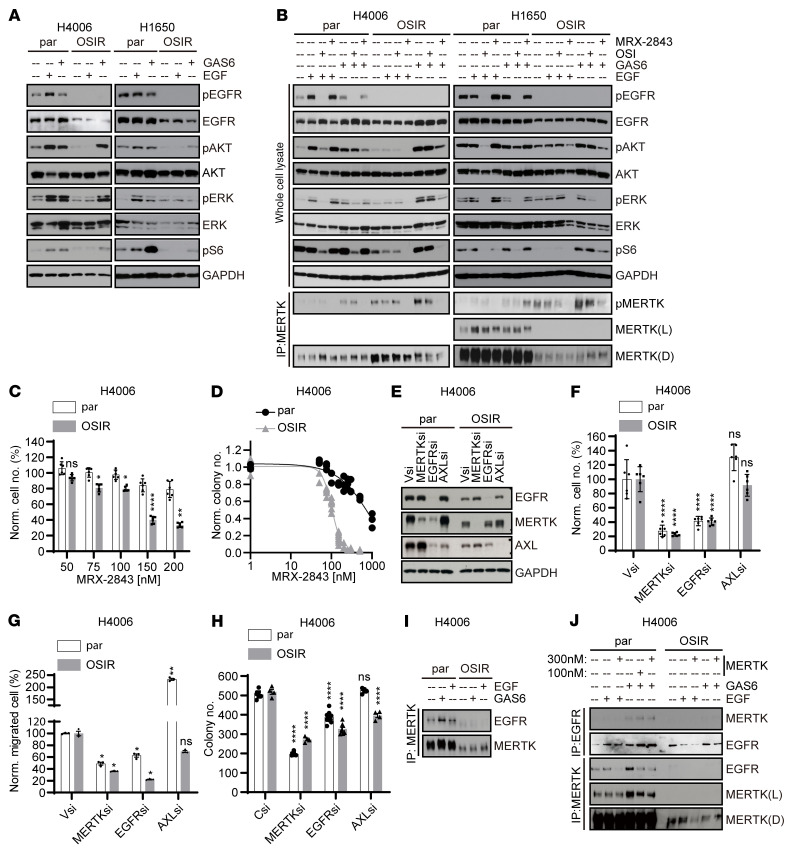
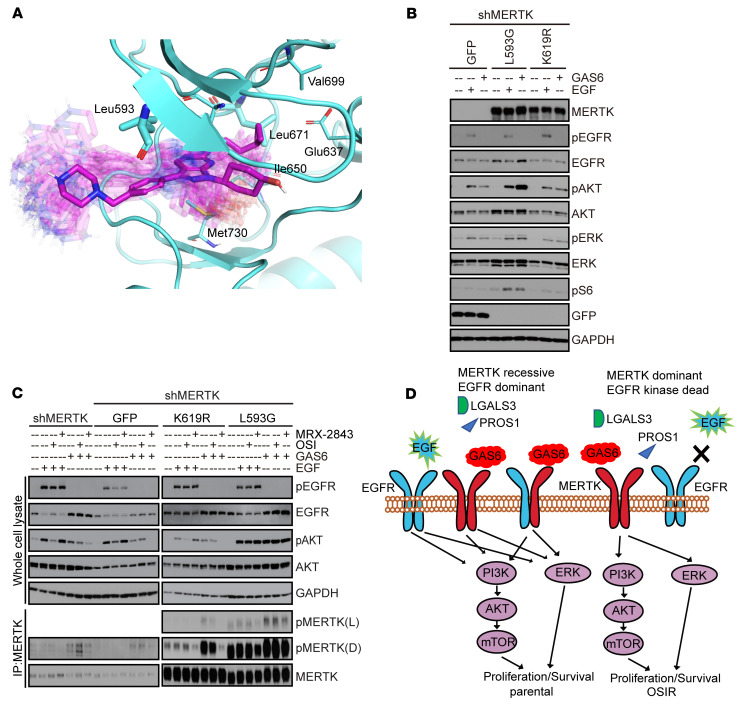
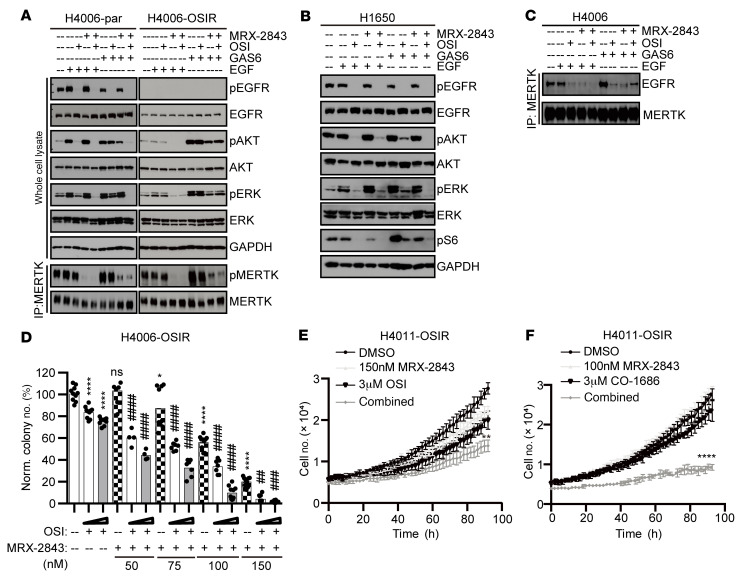
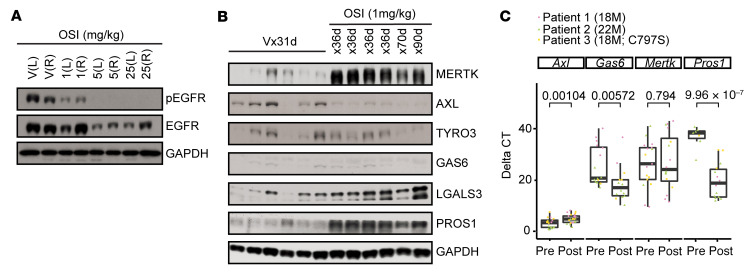
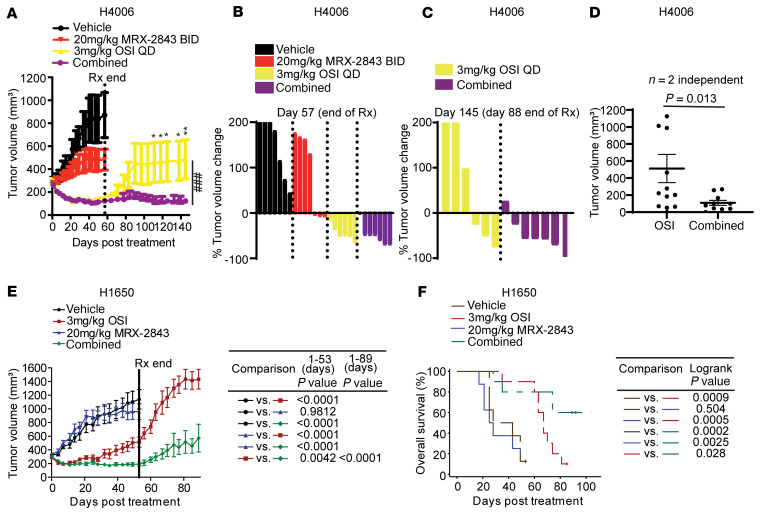
Acquired resistance is inevitable in non-small cell lung cancers (NSCLCs) treated with osimertinib (OSI), and the mechanisms are not well defined. The MERTK ligand GAS6 promoted downstream oncogenic signaling in EGFR-mutated (EGFRMT) NSCLC cells treated with OSI, suggesting a role for MERTK activation in OSI resistance. Indeed, treatment with MRX-2843, a first-in-class MERTK kinase inhibitor, resensitized GAS6-treated NSCLC cells to OSI. Both GAS6 and EGF stimulated downstream PI3K/AKT and MAPK/ERK signaling in parental cells, but only GAS6 activated these pathways in OSI-resistant (OSIR) derivative cell lines. Functionally, OSIR cells were more sensitive to MRX-2843 than parental cells, suggesting acquired dependence on MERTK signaling. Furthermore, MERTK and/or its ligands were dramatically upregulated in EGFRMT tumors after treatment with OSI in both xenograft models and patient samples, consistent with induction of autocrine/paracrine MERTK activation. Moreover, treatment with MRX-2843 in combination with OSI, but not OSI alone, provided durable suppression of tumor growth in vivo, even after treatment was stopped. These data identify MERTK as a driver of bypass signaling in treatment-naive and EGFRMT-OSIR NSCLC cells and predict that MRX-2843 and OSI combination therapy will provide clinical benefit in patients with EGFRMT NSCLC.
Keywords: Cell Biology; Drug therapy; Lung cancer; Oncology; Protein kinases.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
