

RANBP2 and USP9x regulate nuclear import of adenovirus minor coat protein IIIa
- PMID: 35709296
- PMCID: PMC9242475
- DOI: 10.1371/journal.ppat.1010588
RANBP2 and USP9x regulate nuclear import of adenovirus minor coat protein IIIa
Abstract
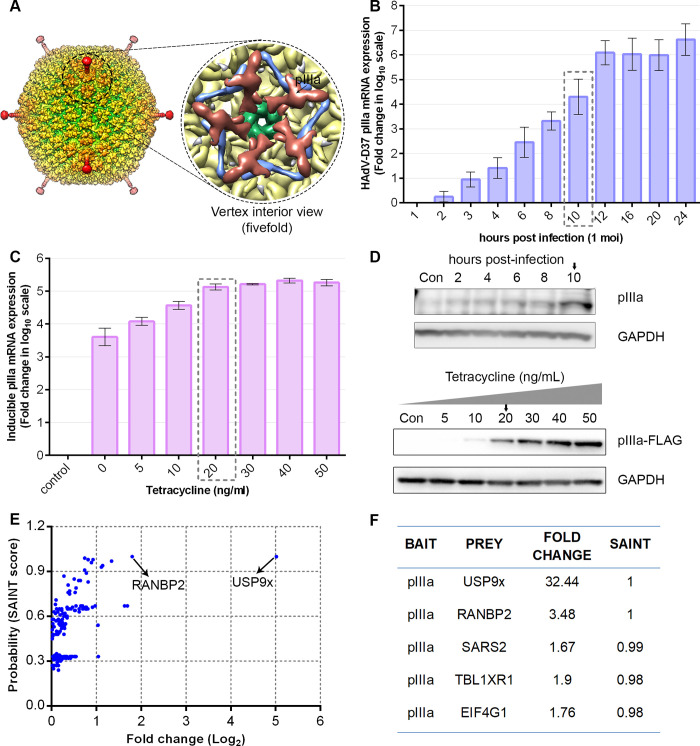
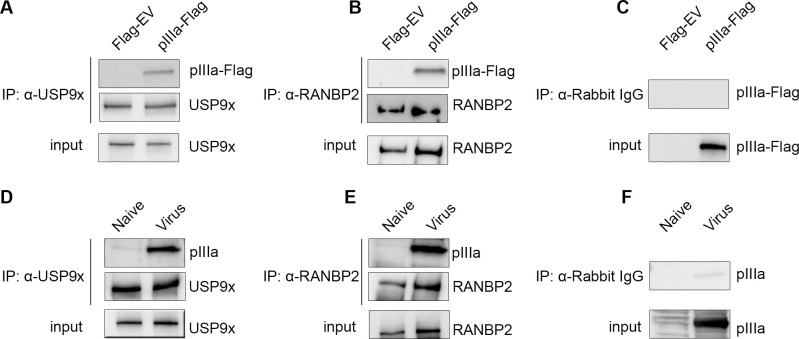
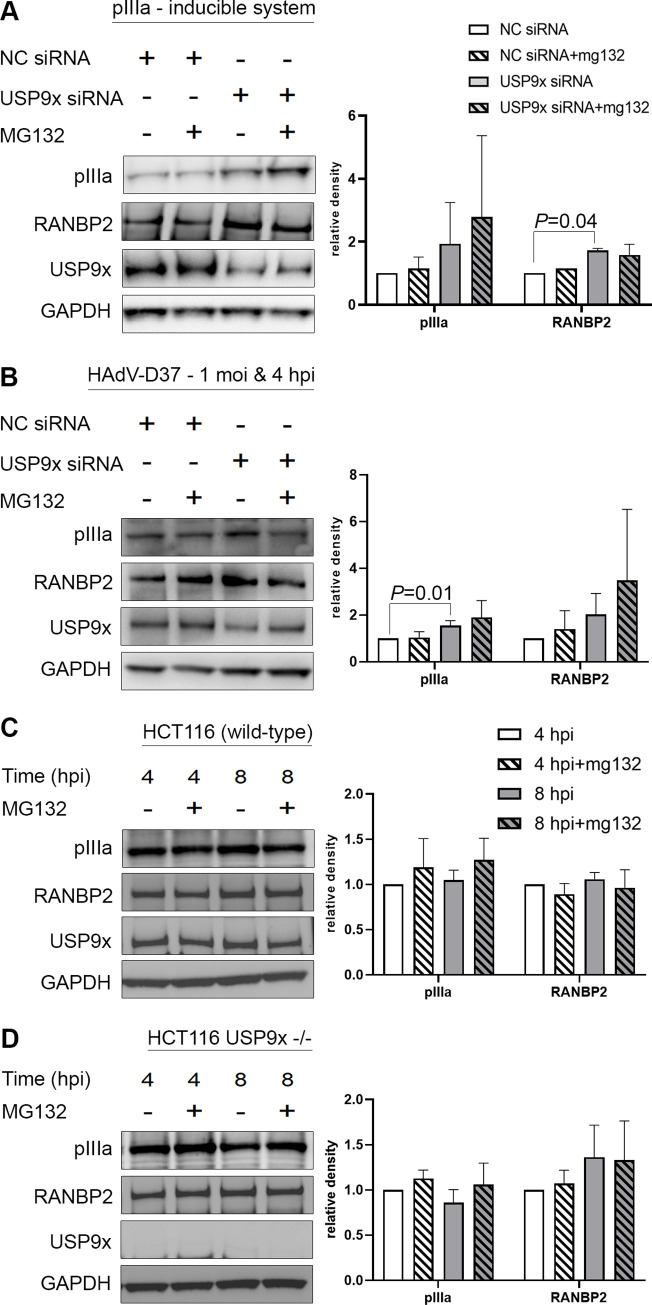
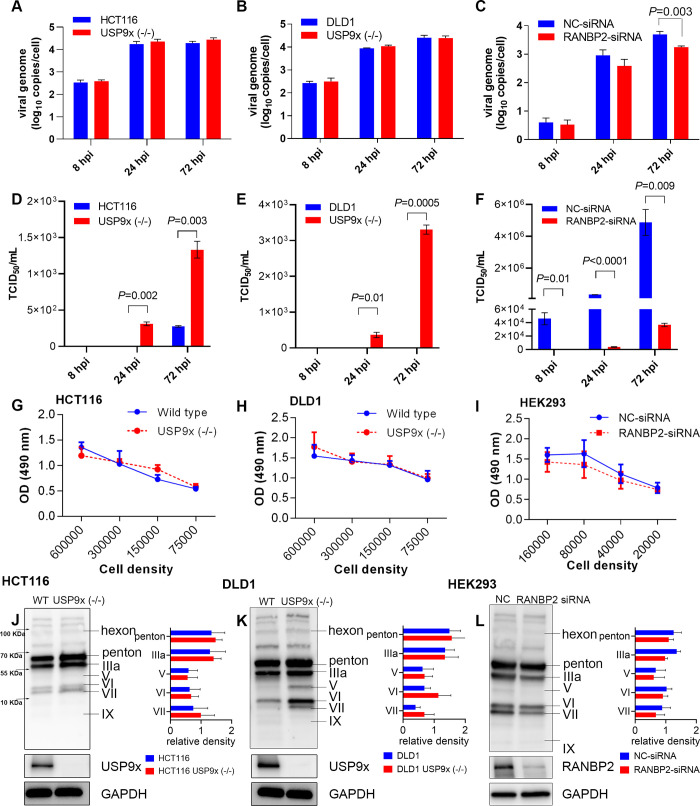
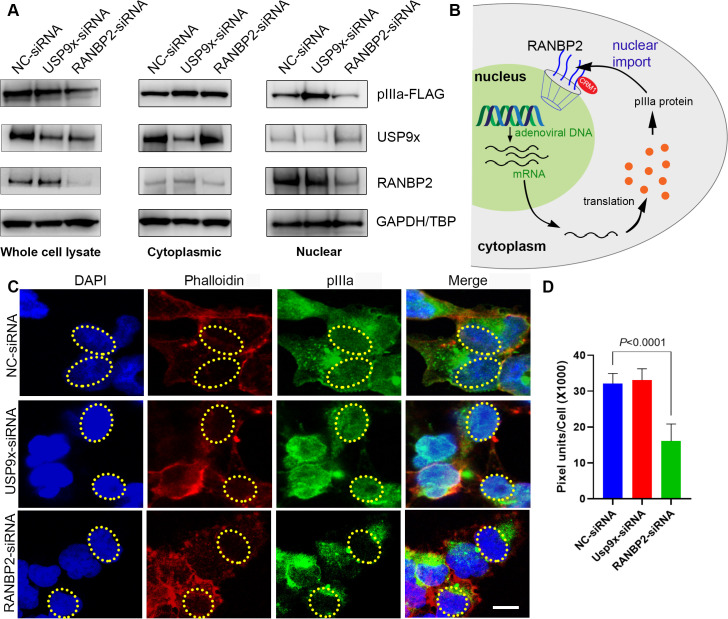
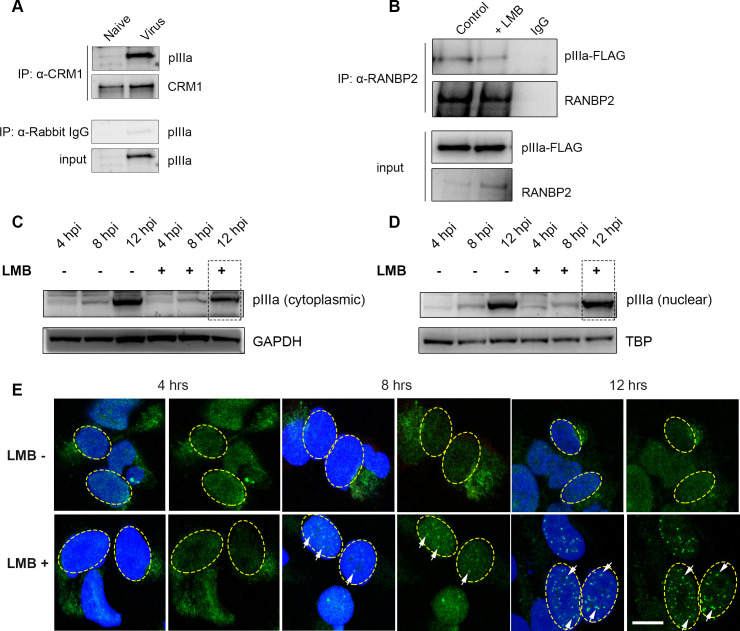
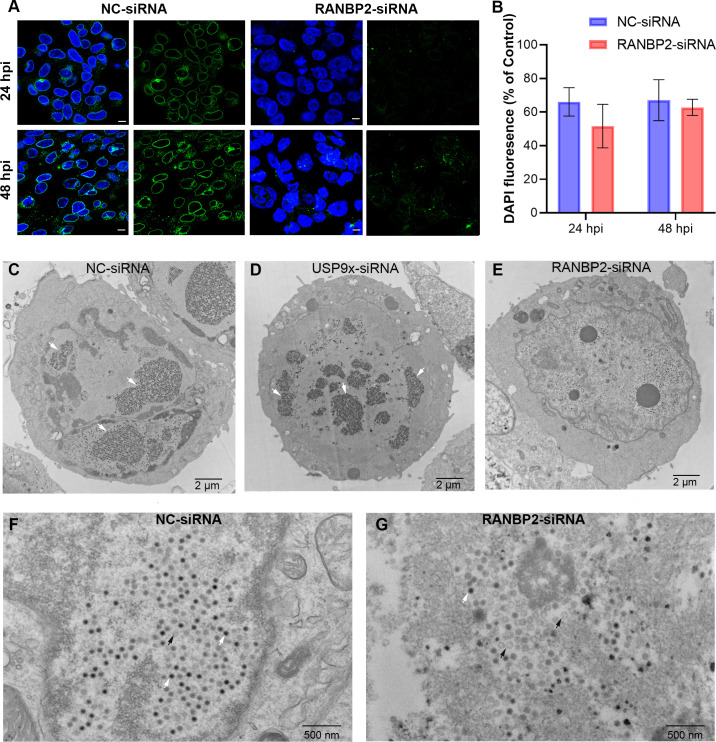
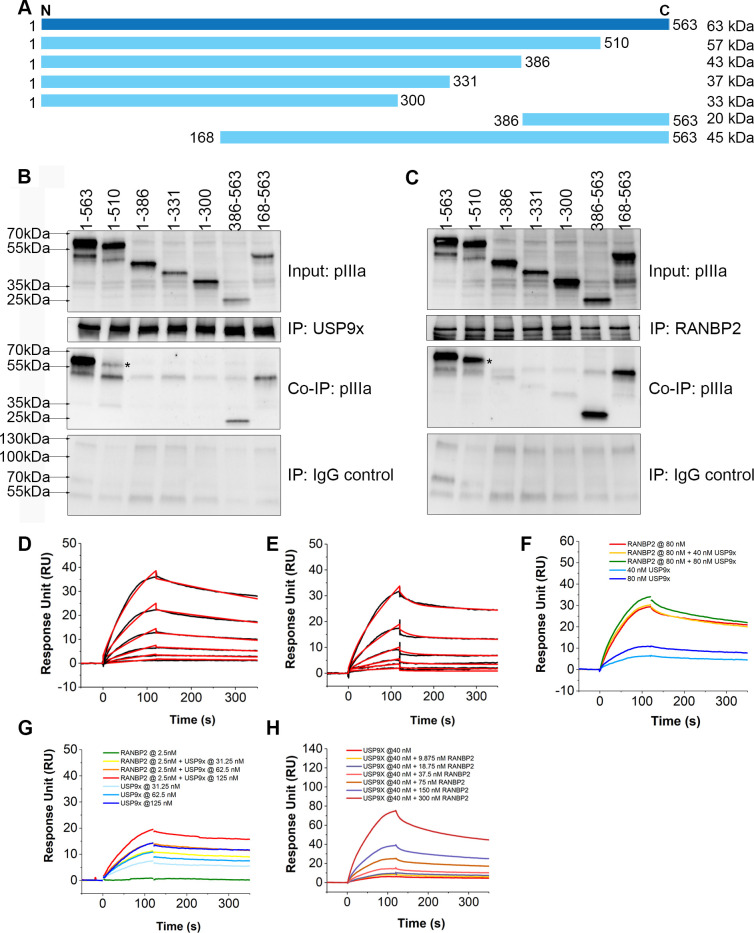
As intracellular parasites, viruses exploit cellular proteins at every stage of infection. Adenovirus outbreaks are associated with severe acute respiratory illnesses and conjunctivitis, with no specific antiviral therapy available. An adenoviral vaccine based on human adenovirus species D (HAdV-D) is currently in use for COVID-19. Herein, we investigate host interactions of HAdV-D type 37 (HAdV-D37) protein IIIa (pIIIa), identified by affinity purification and mass spectrometry (AP-MS) screens. We demonstrate that viral pIIIa interacts with ubiquitin-specific protease 9x (USP9x) and Ran-binding protein 2 (RANBP2). USP9x binding did not invoke its signature deubiquitination function but rather deregulated pIIIa-RANBP2 interactions. In USP9x-knockout cells, viral genome replication and viral protein expression increased compared to wild type cells, supporting a host-favored mechanism for USP9x. Conversely, RANBP2-knock down reduced pIIIa transport to the nucleus, viral genome replication, and viral protein expression. Also, RANBP2-siRNA pretreated cells appeared to contain fewer mature viral particles. Transmission electron microscopy of USP9x-siRNA pretreated, virus-infected cells revealed larger than typical paracrystalline viral arrays. RANBP2-siRNA pretreatment led to the accumulation of defective assembly products at an early maturation stage. CRM1 nuclear export blockade by leptomycin B led to the retention of pIIIa within cell nuclei and hindered pIIIa-RANBP2 interactions. In-vitro binding analyses indicated that USP9x and RANBP2 bind to C-terminus of pIIIa amino acids 386-563 and 386-510, respectively. Surface plasmon resonance testing showed direct pIIIa interaction with recombinant USP9x and RANBP2 proteins, without competition. Using an alternative and genetically disparate adenovirus type (HAdV-C5), we show that the demonstrated pIIIa interaction is also important for a severe respiratory pathogen. Together, our results suggest that pIIIa hijacks RANBP2 for nuclear import and subsequent virion assembly. USP9x counteracts this interaction and negatively regulates virion synthesis. This analysis extends the scope of known adenovirus-host interactions and has potential implications in designing new antiviral therapeutics.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
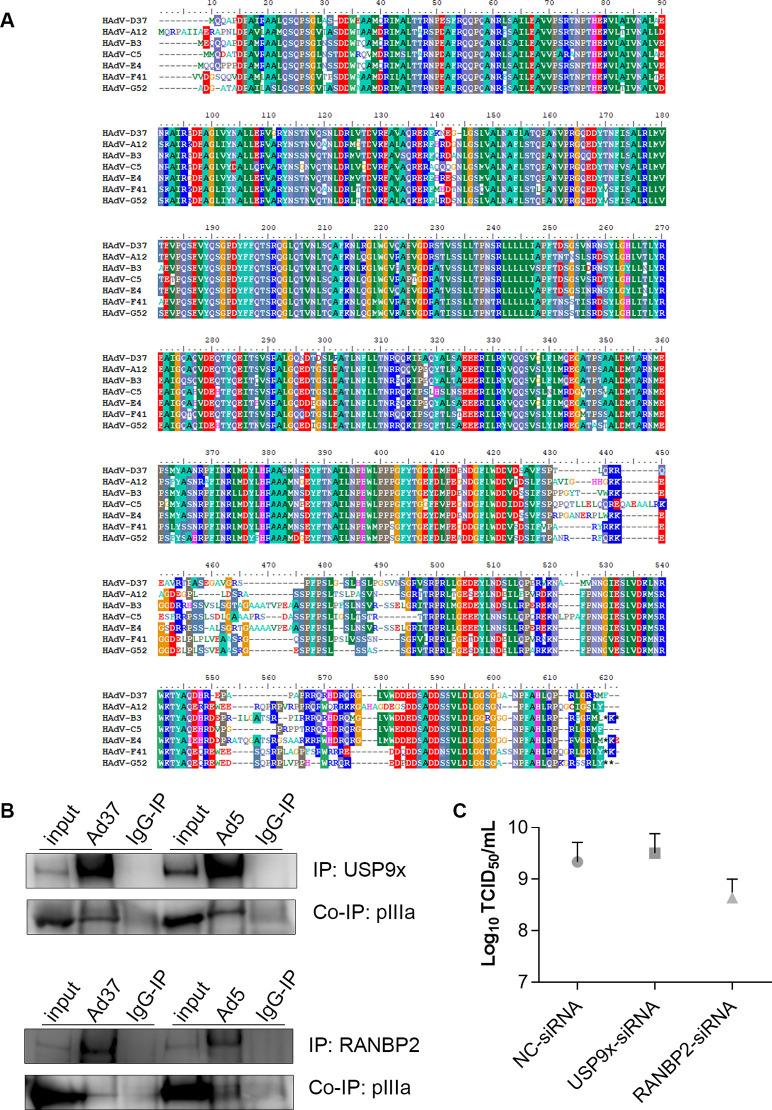
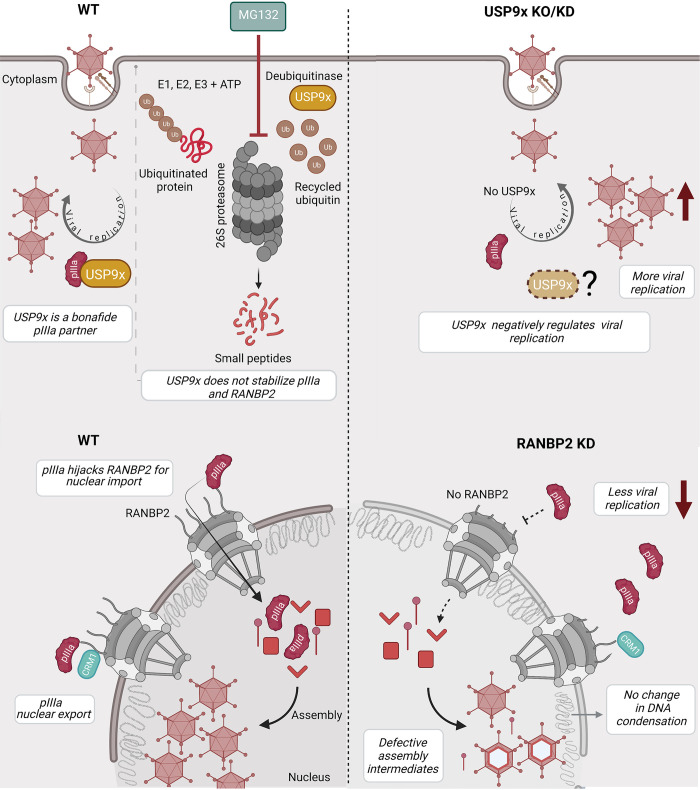
References
-
- Hernandez Duran A, Greco TM, Vollmer B, Cristea IM, Grunewald K, Topf M. Protein interactions and consensus clustering analysis uncover insights into herpesvirus virion structure and function relationships. PLoS Biol. 2019;17(6):e3000316. doi: 10.1371/journal.pbio.3000316 ; PubMed Central PMCID: PMC6594648. - DOI - PMC - PubMed
-
- Martinez-Martin N, Ramani SR, Hackney JA, Tom I, Wranik BJ, Chan M, et al. The extracellular interactome of the human adenovirus family reveals diverse strategies for immunomodulation. Nat Commun. 2016;7:11473. Epub 2016/05/06. doi: 10.1038/ncomms11473 ; PubMed Central PMCID: PMC4858740. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
