

Multi-view heterogeneous molecular network representation learning for protein-protein interaction prediction
- PMID: 35710342
- PMCID: PMC9205098
- DOI: 10.1186/s12859-022-04766-z
Multi-view heterogeneous molecular network representation learning for protein-protein interaction prediction
Abstract
Background: Protein-protein interaction (PPI) plays an important role in regulating cells and signals. Despite the ongoing efforts of the bioassay group, continued incomplete data limits our ability to understand the molecular roots of human disease. Therefore, it is urgent to develop a computational method to predict PPIs from the perspective of molecular system.
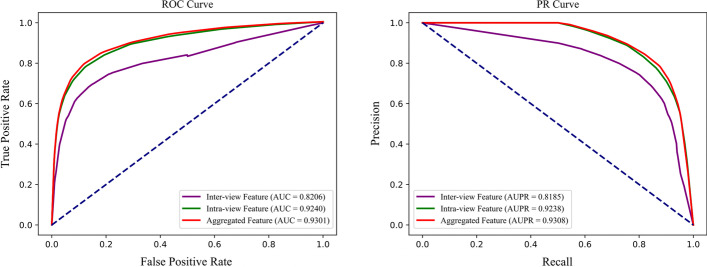
Methods: In this paper, a highly efficient computational model, MTV-PPI, is proposed for PPI prediction based on a heterogeneous molecular network by learning inter-view protein sequences and intra-view interactions between molecules simultaneously. On the one hand, the inter-view feature is extracted from the protein sequence by k-mer method. On the other hand, we use a popular embedding method LINE to encode the heterogeneous molecular network to obtain the intra-view feature. Thus, the protein representation used in MTV-PPI is constructed by the aggregation of its inter-view feature and intra-view feature. Finally, random forest is integrated to predict potential PPIs.
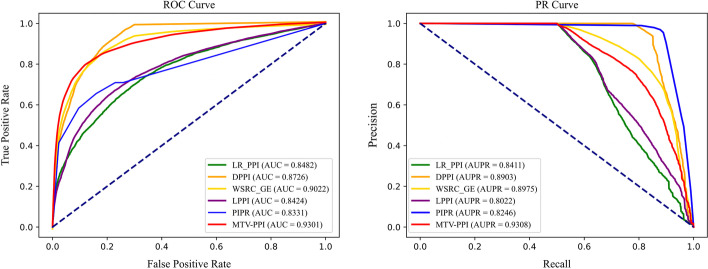
Results: To prove the effectiveness of MTV-PPI, we conduct extensive experiments on a collected heterogeneous molecular network with the accuracy of 86.55%, sensitivity of 82.49%, precision of 89.79%, AUC of 0.9301 and AUPR of 0.9308. Further comparison experiments are performed with various protein representations and classifiers to indicate the effectiveness of MTV-PPI in predicting PPIs based on a complex network.
Conclusion: The achieved experimental results illustrate that MTV-PPI is a promising tool for PPI prediction, which may provide a new perspective for the future interactions prediction researches based on heterogeneous molecular network.
Keywords: Heterogeneous molecular network; LINE; Network representation learning; Protein sequence; Protein–protein interaction.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures






Similar articles
-
SDNN-PPI: self-attention with deep neural network effect on protein-protein interaction prediction.BMC Genomics. 2022 Jun 27;23(1):474. doi: 10.1186/s12864-022-08687-2. BMC Genomics. 2022. PMID: 35761175 Free PMC article.
-
Predicting protein-protein interactions from primary protein sequences using a novel multi-scale local feature representation scheme and the random forest.PLoS One. 2015 May 6;10(5):e0125811. doi: 10.1371/journal.pone.0125811. eCollection 2015. PLoS One. 2015. PMID: 25946106 Free PMC article.
-
DSSGNN-PPI: A Protein-Protein Interactions prediction model based on Double Structure and Sequence graph neural networks.Comput Biol Med. 2024 Jul;177:108669. doi: 10.1016/j.compbiomed.2024.108669. Epub 2024 May 29. Comput Biol Med. 2024. PMID: 38833802
-
Machine learning on protein-protein interaction prediction: models, challenges and trends.Brief Bioinform. 2023 Mar 19;24(2):bbad076. doi: 10.1093/bib/bbad076. Brief Bioinform. 2023. PMID: 36880207 Review.
-
Application of Machine Learning Approaches for Protein-protein Interactions Prediction.Med Chem. 2017;13(6):506-514. doi: 10.2174/1573406413666170522150940. Med Chem. 2017. PMID: 28530547 Review.
Cited by
-
Intelligent Protein Design and Molecular Characterization Techniques: A Comprehensive Review.Molecules. 2023 Nov 30;28(23):7865. doi: 10.3390/molecules28237865. Molecules. 2023. PMID: 38067593 Free PMC article. Review.
-
Effectively predicting HIV-1 protease cleavage sites by using an ensemble learning approach.BMC Bioinformatics. 2022 Oct 27;23(1):447. doi: 10.1186/s12859-022-04999-y. BMC Bioinformatics. 2022. PMID: 36303135 Free PMC article.
-
GNNGL-PPI: multi-category prediction of protein-protein interactions using graph neural networks based on global graphs and local subgraphs.BMC Genomics. 2024 May 9;25(1):406. doi: 10.1186/s12864-024-10299-x. BMC Genomics. 2024. PMID: 38724906 Free PMC article.
-
DNA sequence analysis landscape: a comprehensive review of DNA sequence analysis task types, databases, datasets, word embedding methods, and language models.Front Med (Lausanne). 2025 Apr 8;12:1503229. doi: 10.3389/fmed.2025.1503229. eCollection 2025. Front Med (Lausanne). 2025. PMID: 40265190 Free PMC article. Review.
-
Prediction of protein interactions with function in protein (de-)phosphorylation.PLoS One. 2025 Mar 3;20(3):e0319084. doi: 10.1371/journal.pone.0319084. eCollection 2025. PLoS One. 2025. PMID: 40029919 Free PMC article.
References
-
- Luo X, Ming Z, You Z, Li S, Xia Y, Leung H. Improving network topology-based protein interactome mapping via collaborative filtering. Knowl Based Syst. 2015;90:23–32. doi: 10.1016/j.knosys.2015.10.003. - DOI
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
