

Disentangling the genetic basis of rhizosphere microbiome assembly in tomato
- PMID: 35710629
- PMCID: PMC9203511
- DOI: 10.1038/s41467-022-30849-9
Disentangling the genetic basis of rhizosphere microbiome assembly in tomato
Abstract
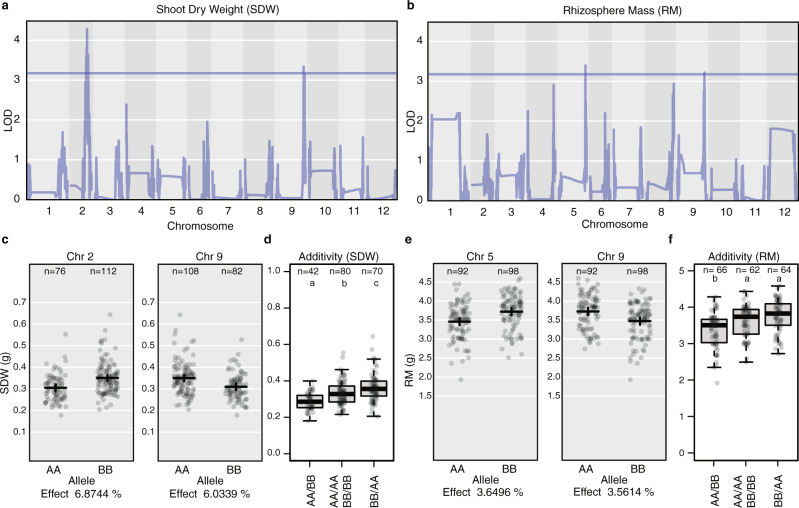
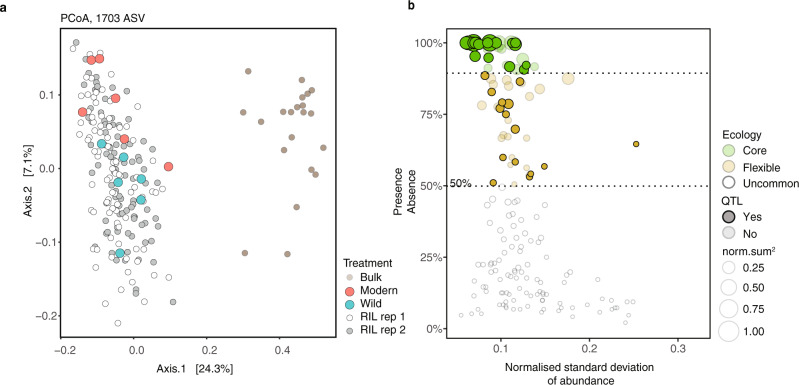
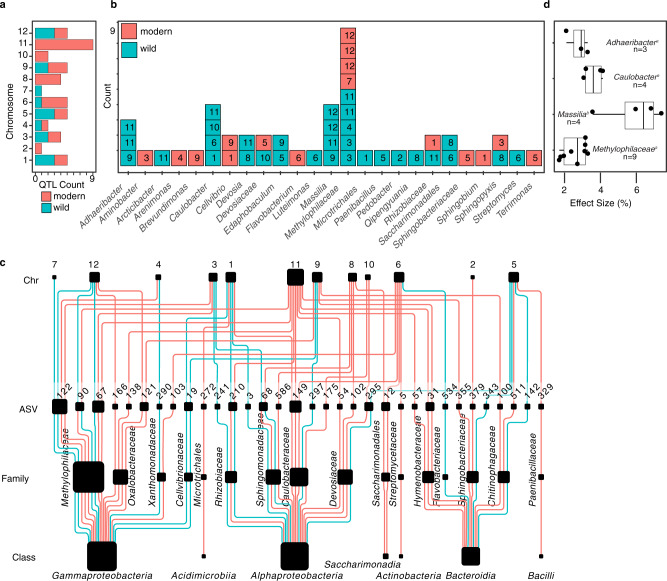
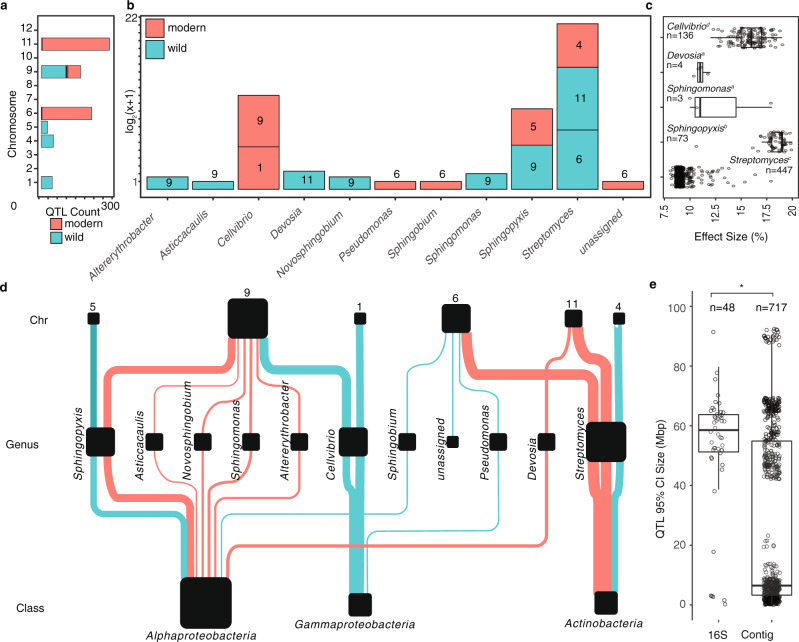
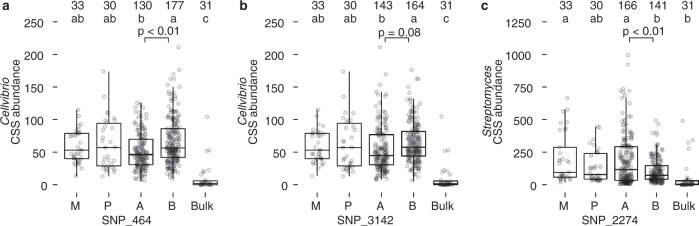
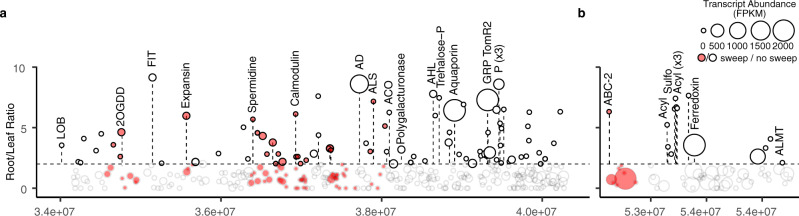
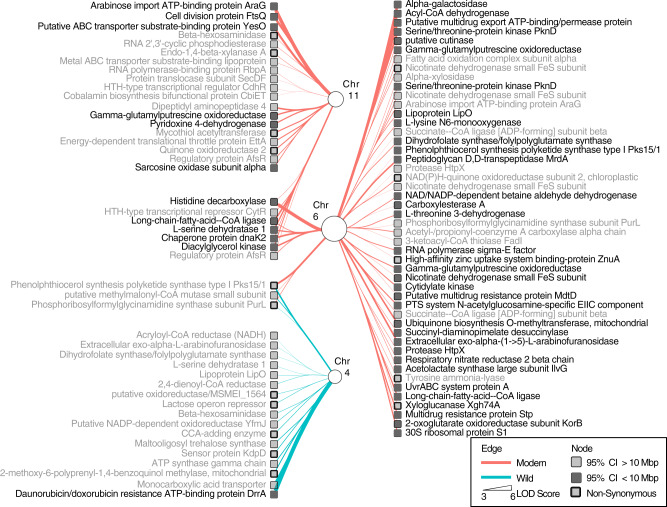
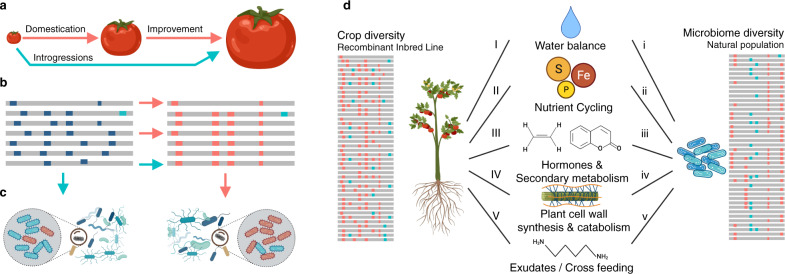
Microbiomes play a pivotal role in plant growth and health, but the genetic factors involved in microbiome assembly remain largely elusive. Here, we map the molecular features of the rhizosphere microbiome as quantitative traits of a diverse hybrid population of wild and domesticated tomato. Gene content analysis of prioritized tomato quantitative trait loci suggests a genetic basis for differential recruitment of various rhizobacterial lineages, including a Streptomyces-associated 6.31 Mbp region harboring tomato domestication sweeps and encoding, among others, the iron regulator FIT and the water channel aquaporin SlTIP2.3. Within metagenome-assembled genomes of root-associated Streptomyces and Cellvibrio, we identify bacterial genes involved in metabolism of plant polysaccharides, iron, sulfur, trehalose, and vitamins, whose genetic variation associates with specific tomato QTLs. By integrating 'microbiomics' and quantitative plant genetics, we pinpoint putative plant and reciprocal rhizobacterial traits underlying microbiome assembly, thereby providing a first step towards plant-microbiome breeding programs.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
