

Epigenomic and transcriptomic analyses define core cell types, genes and targetable mechanisms for kidney disease
- PMID: 35710981
- PMCID: PMC11626562
- DOI: 10.1038/s41588-022-01097-w
Epigenomic and transcriptomic analyses define core cell types, genes and targetable mechanisms for kidney disease
Abstract
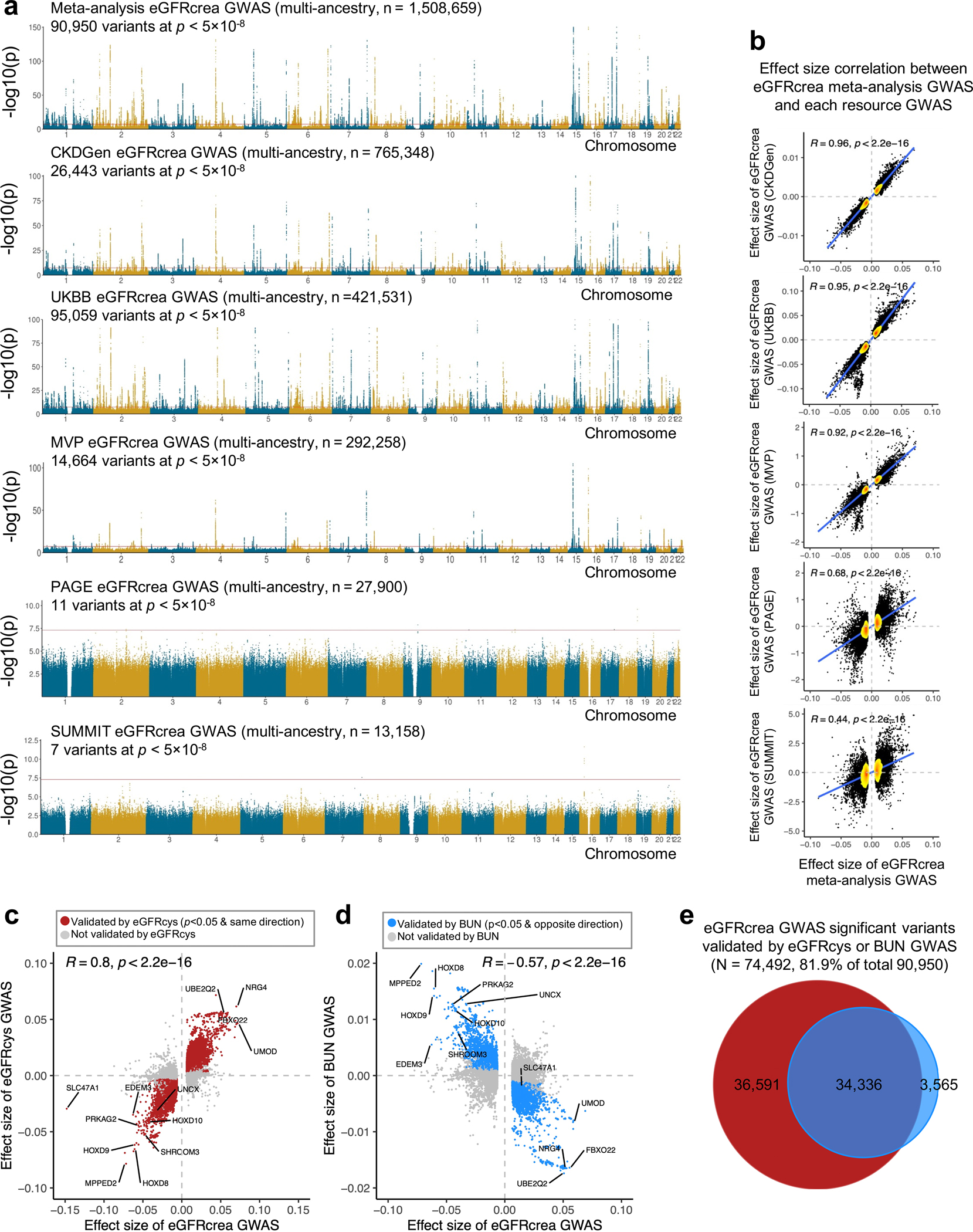
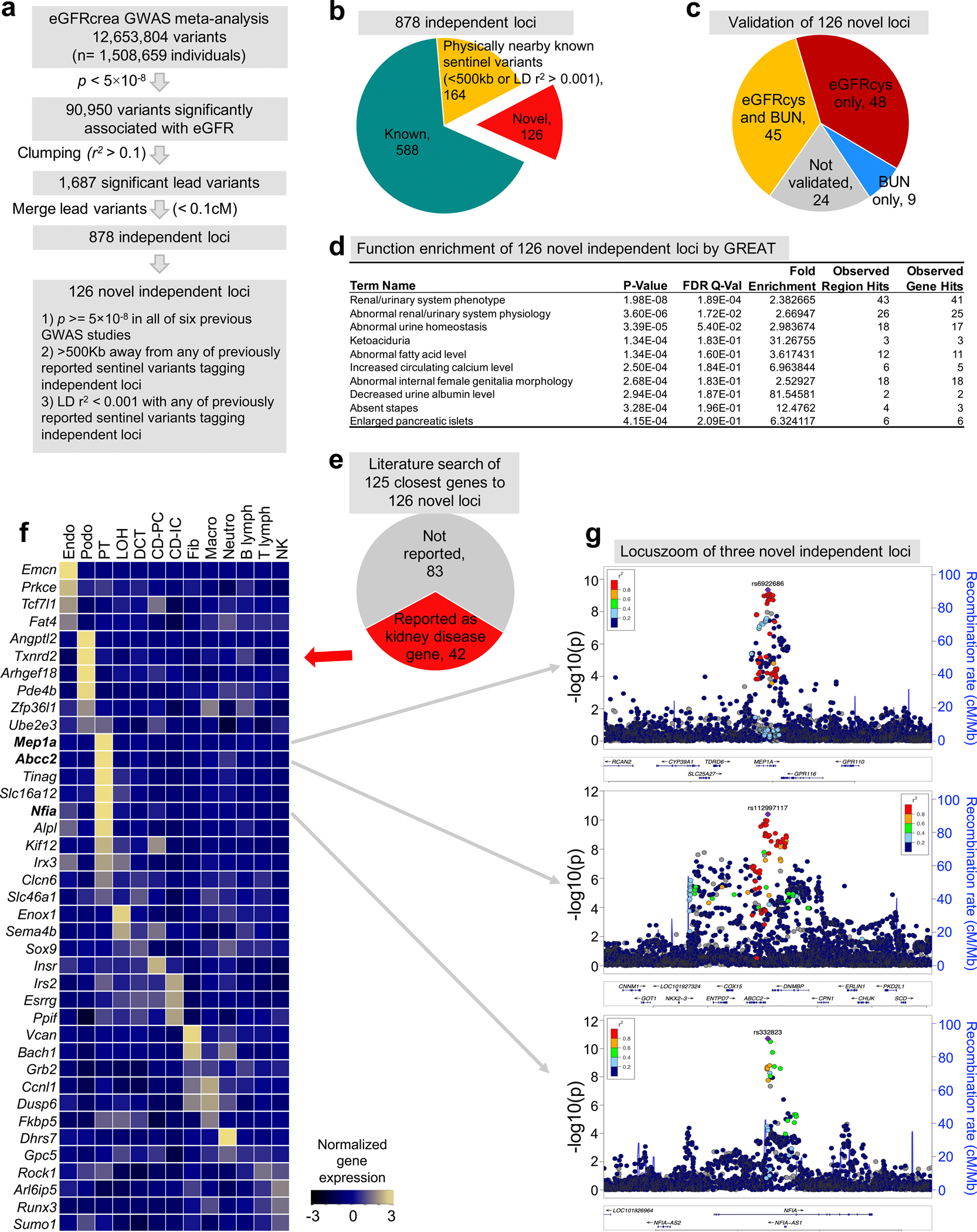
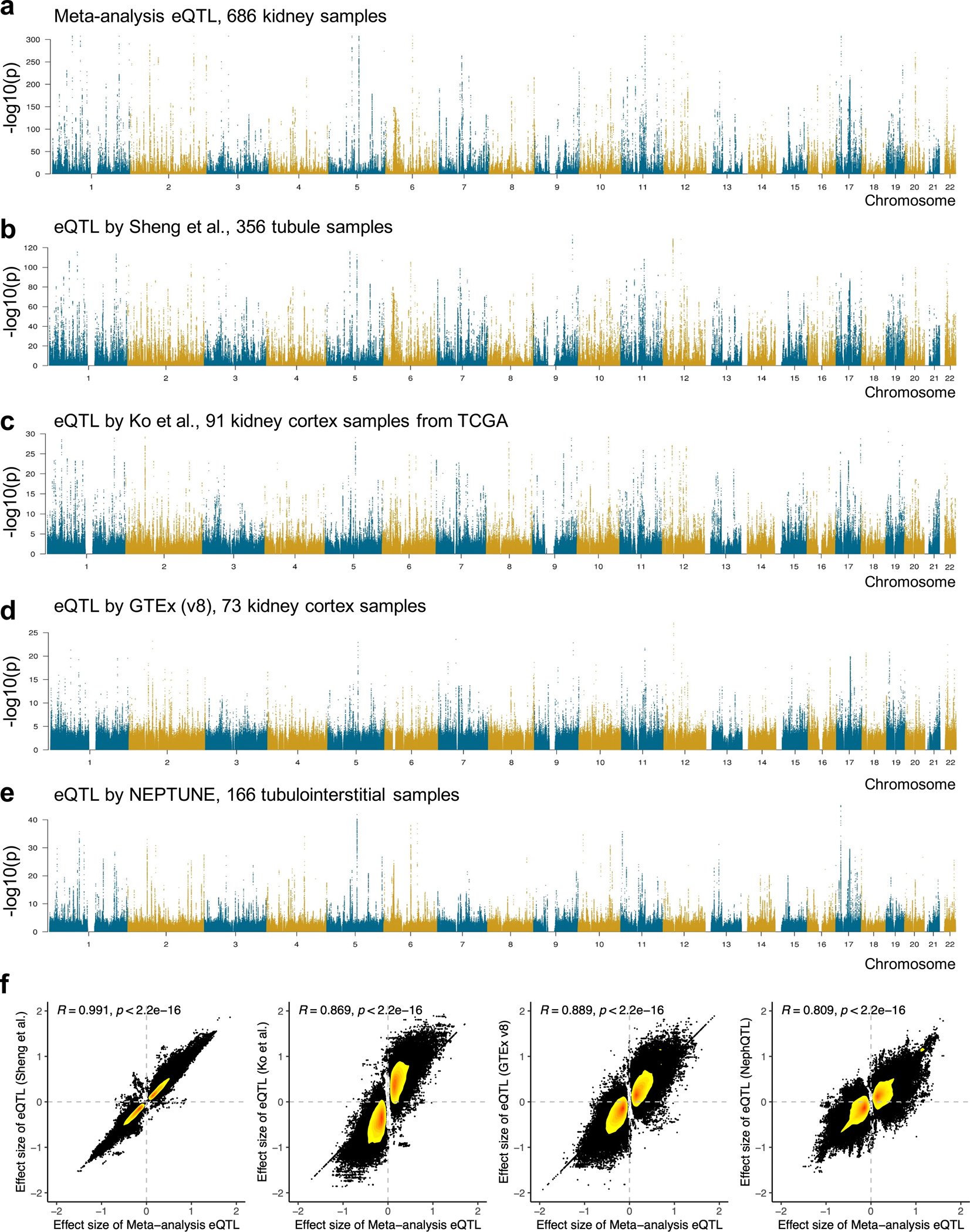
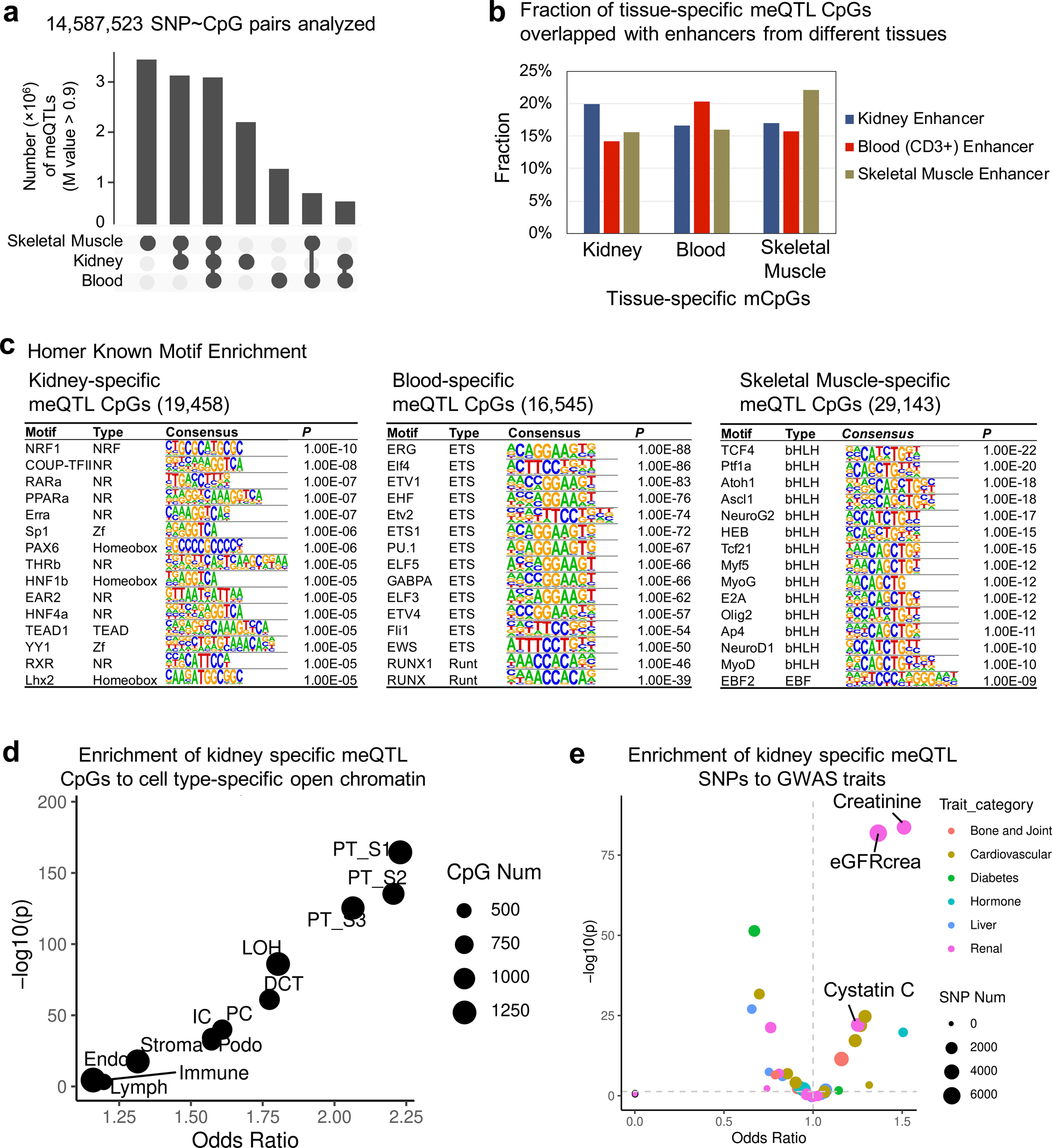
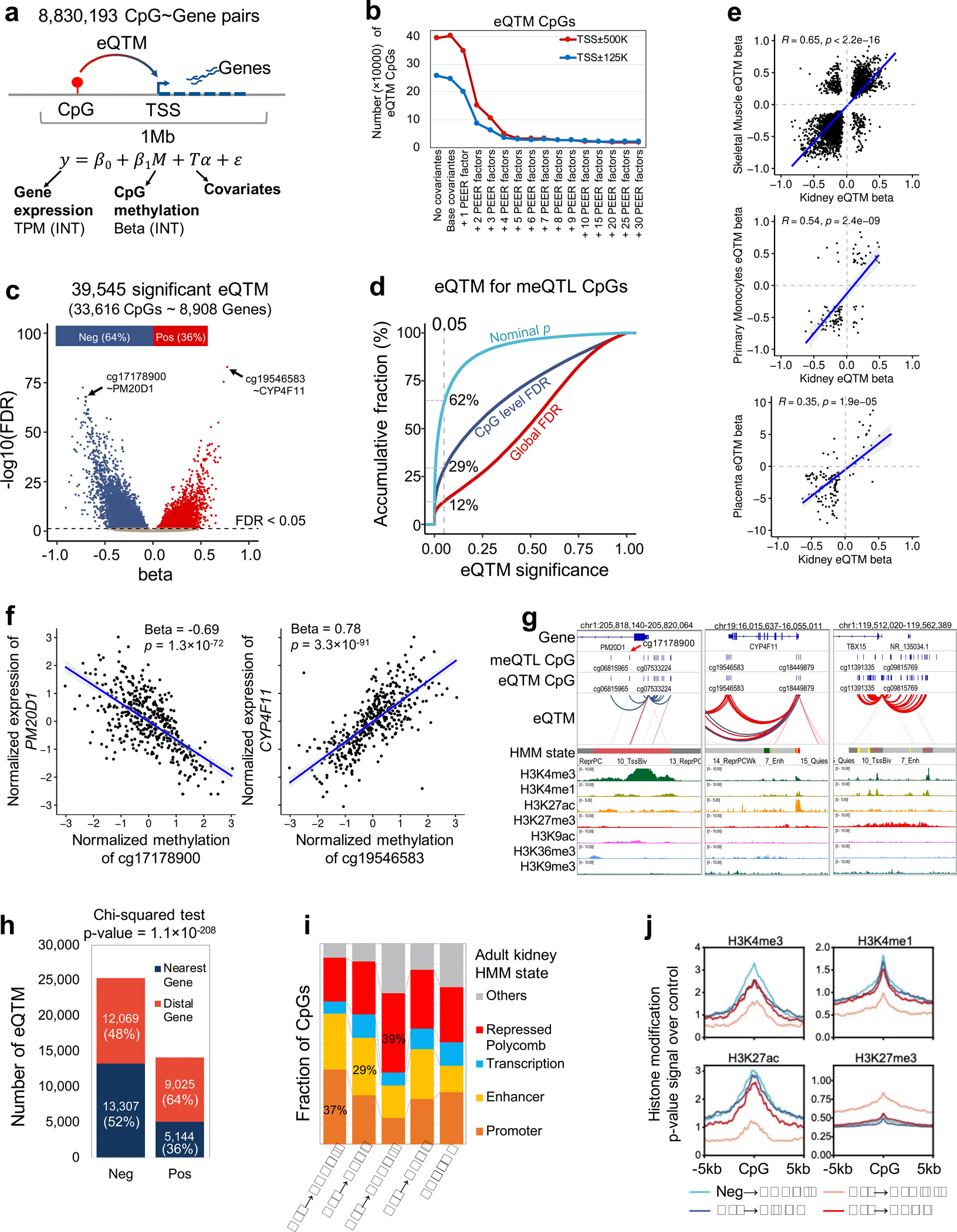
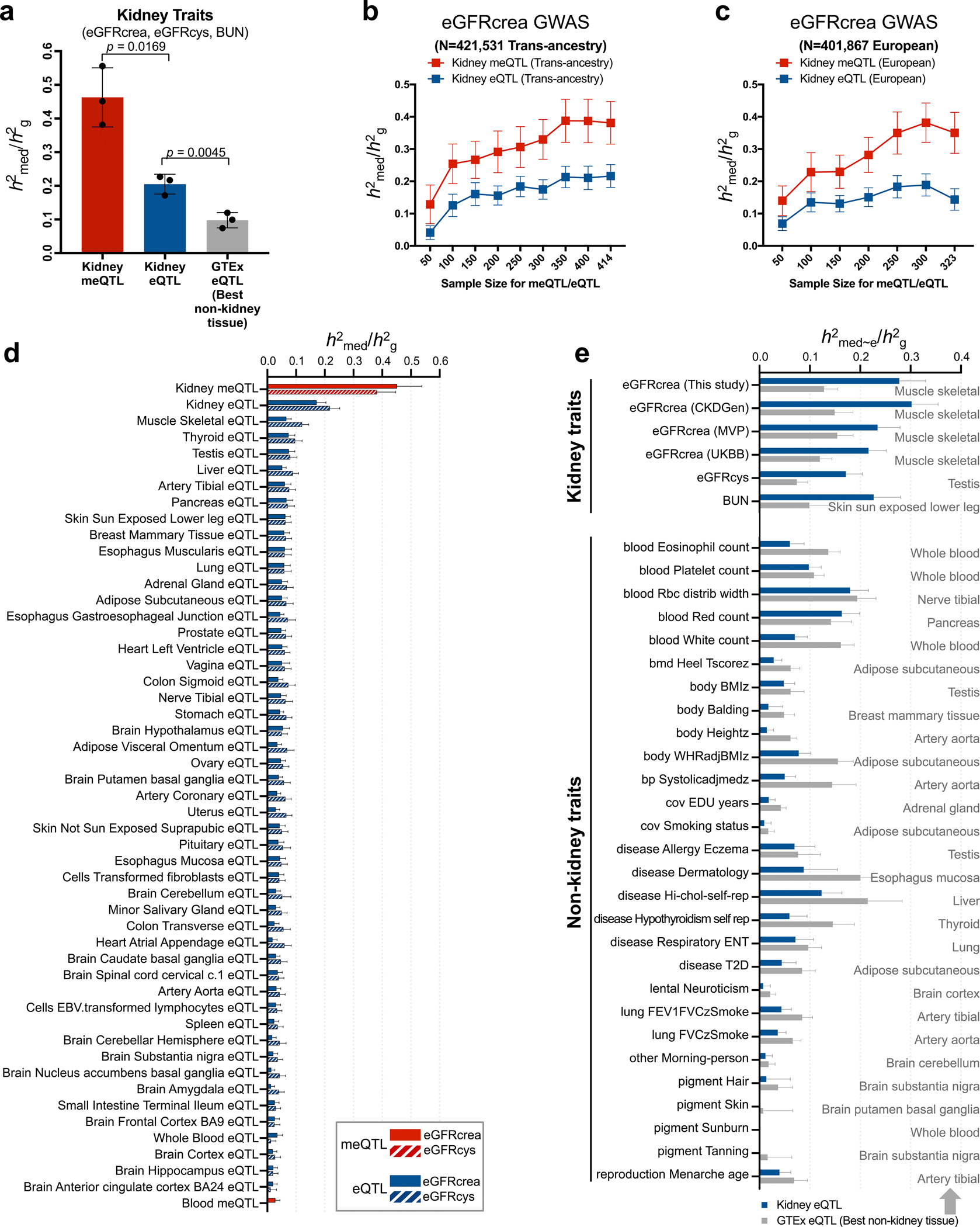
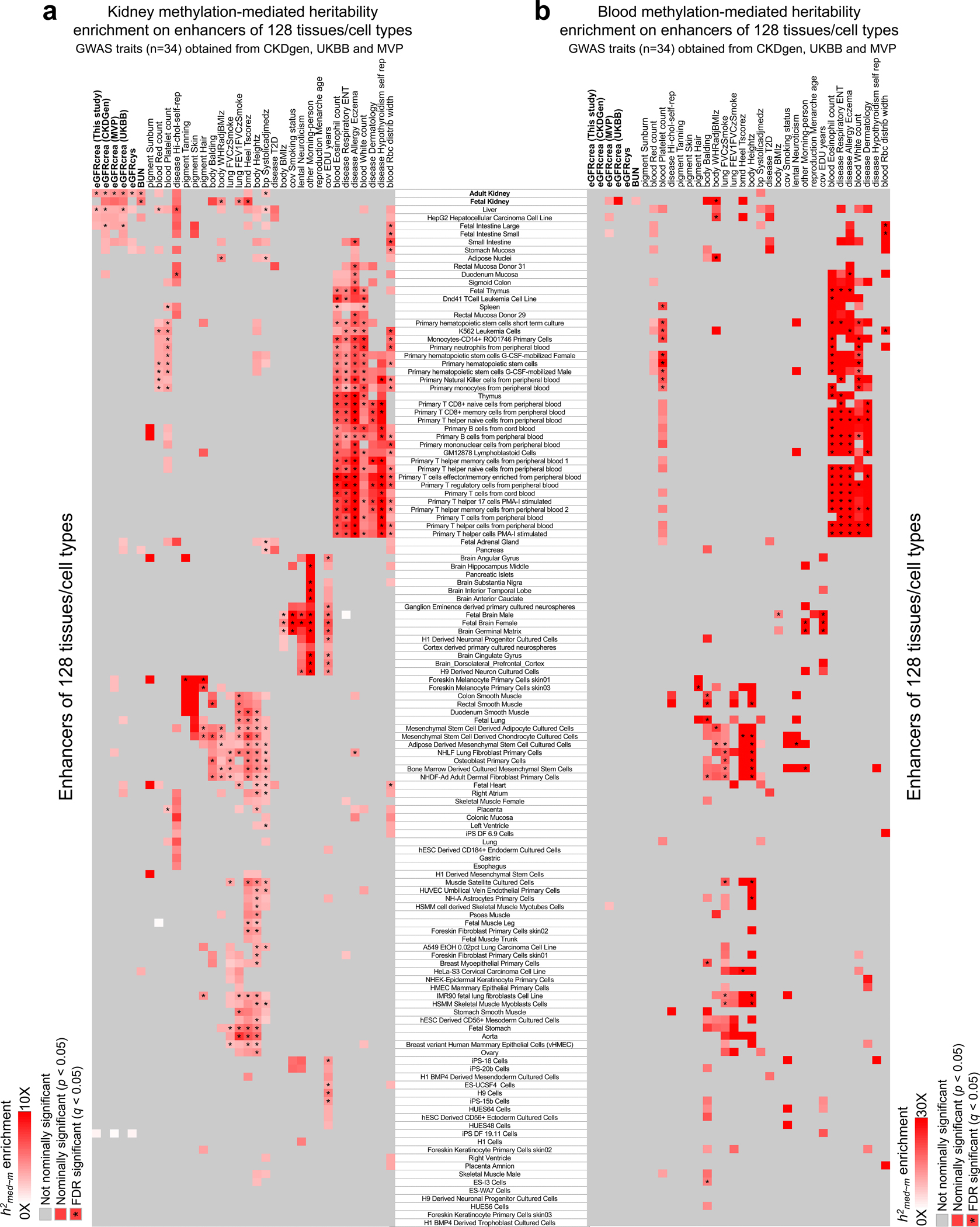
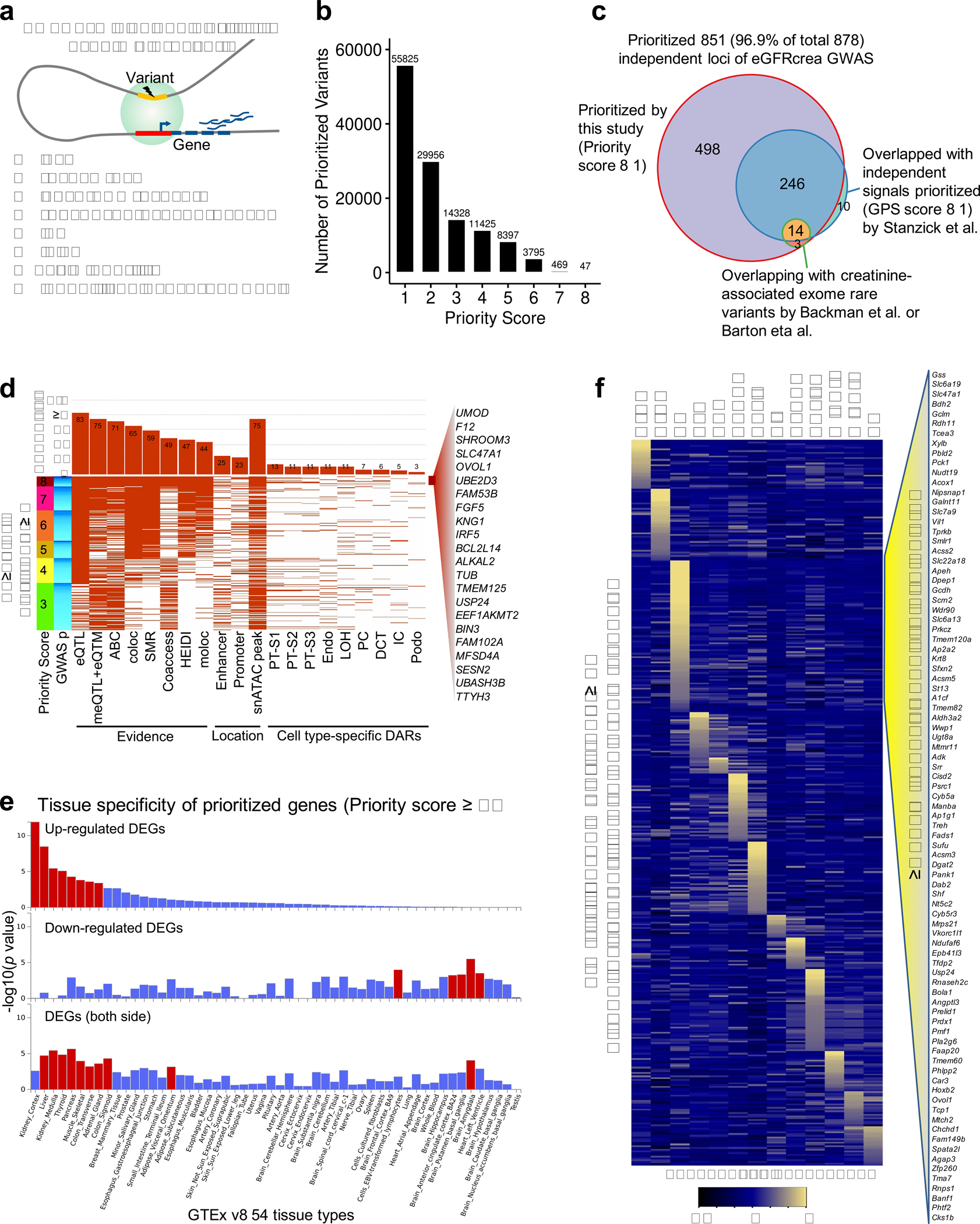
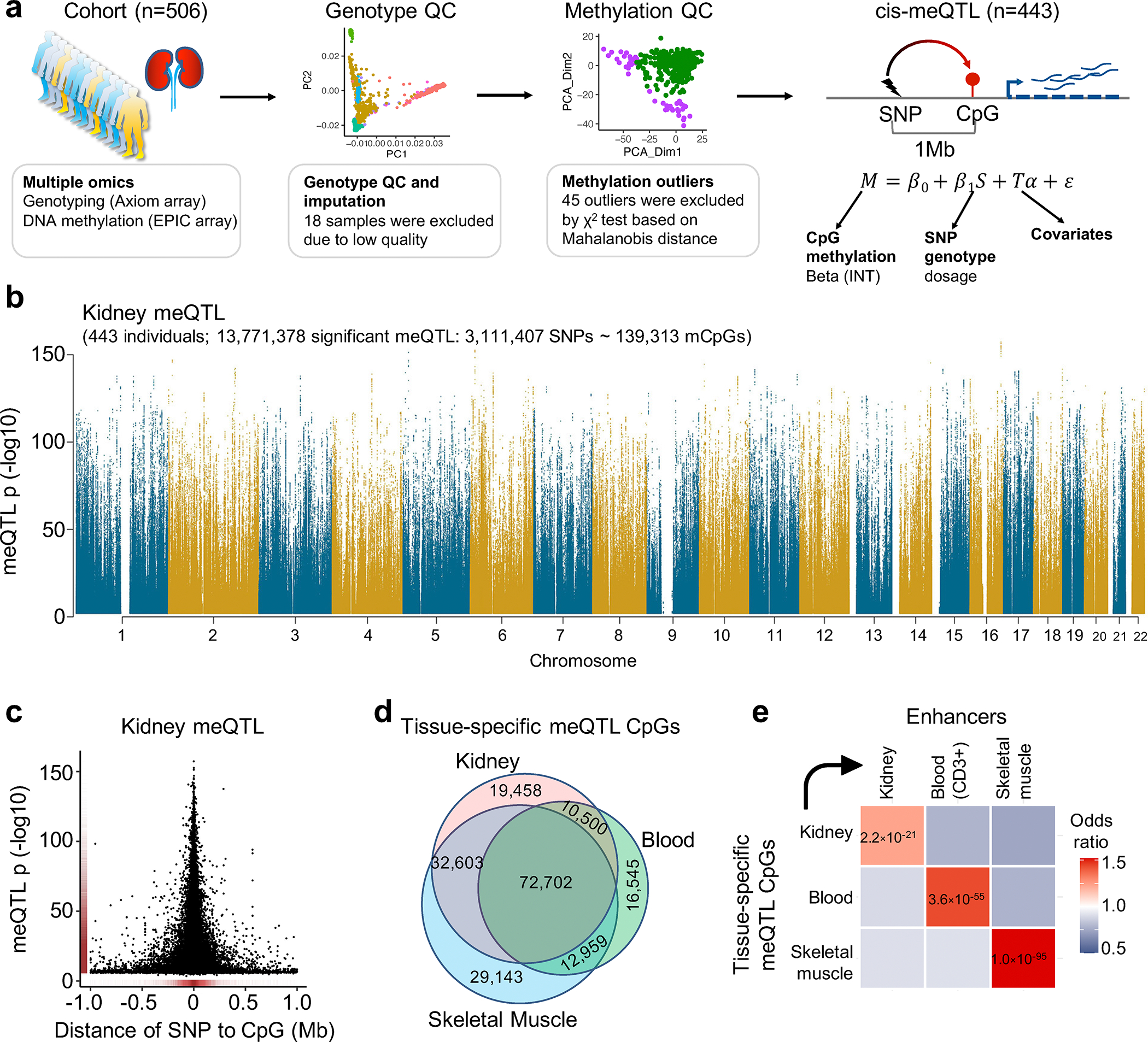
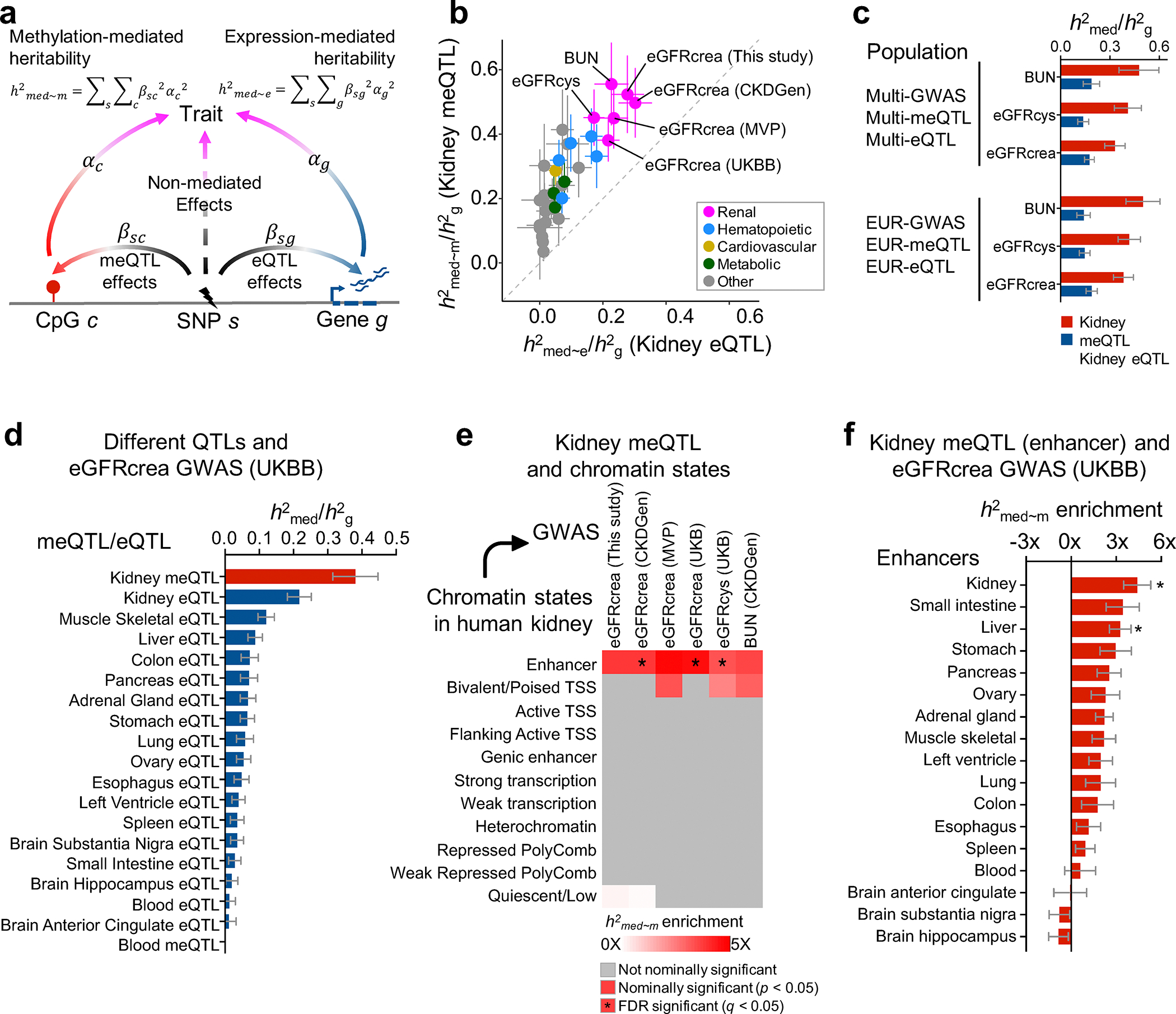
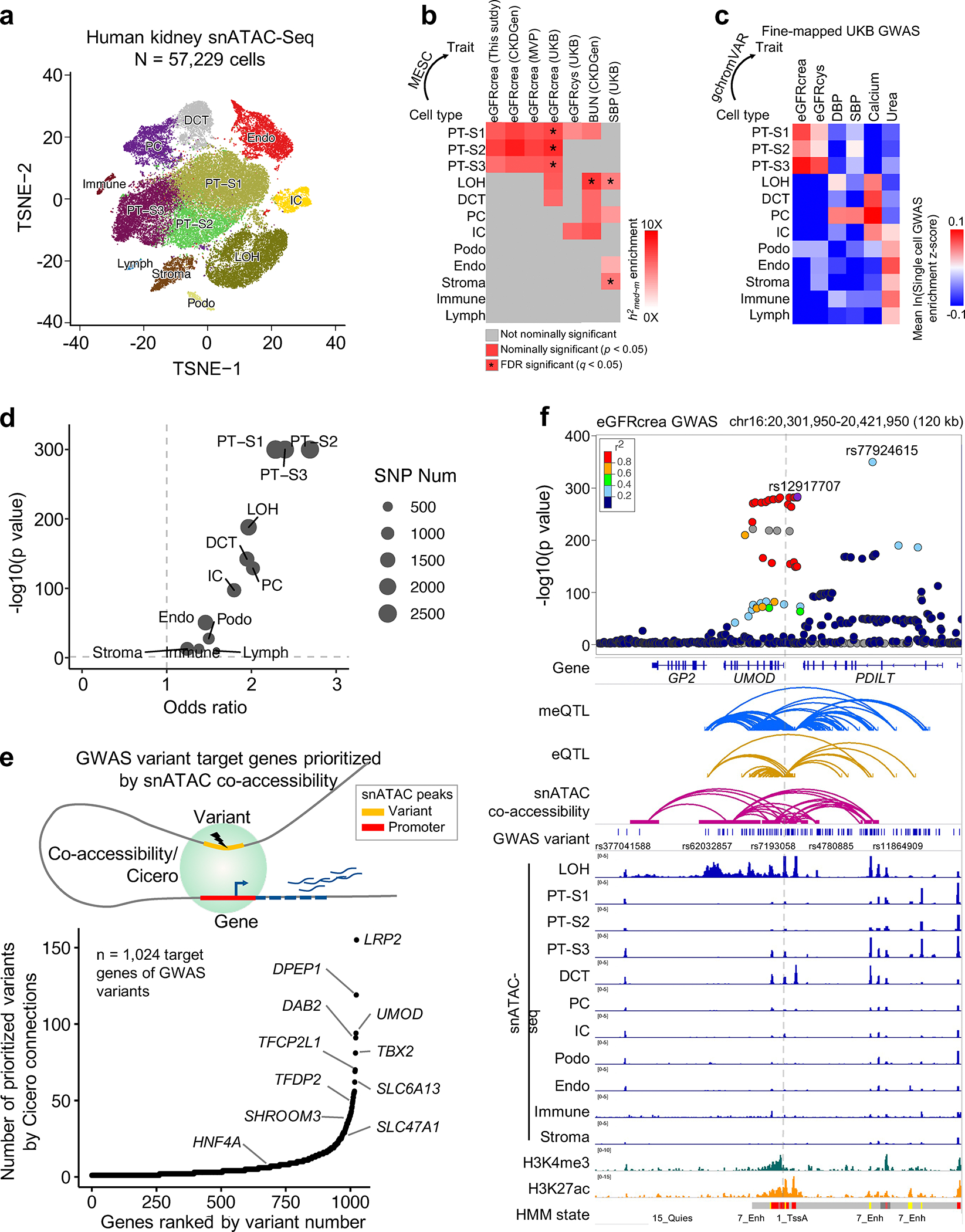
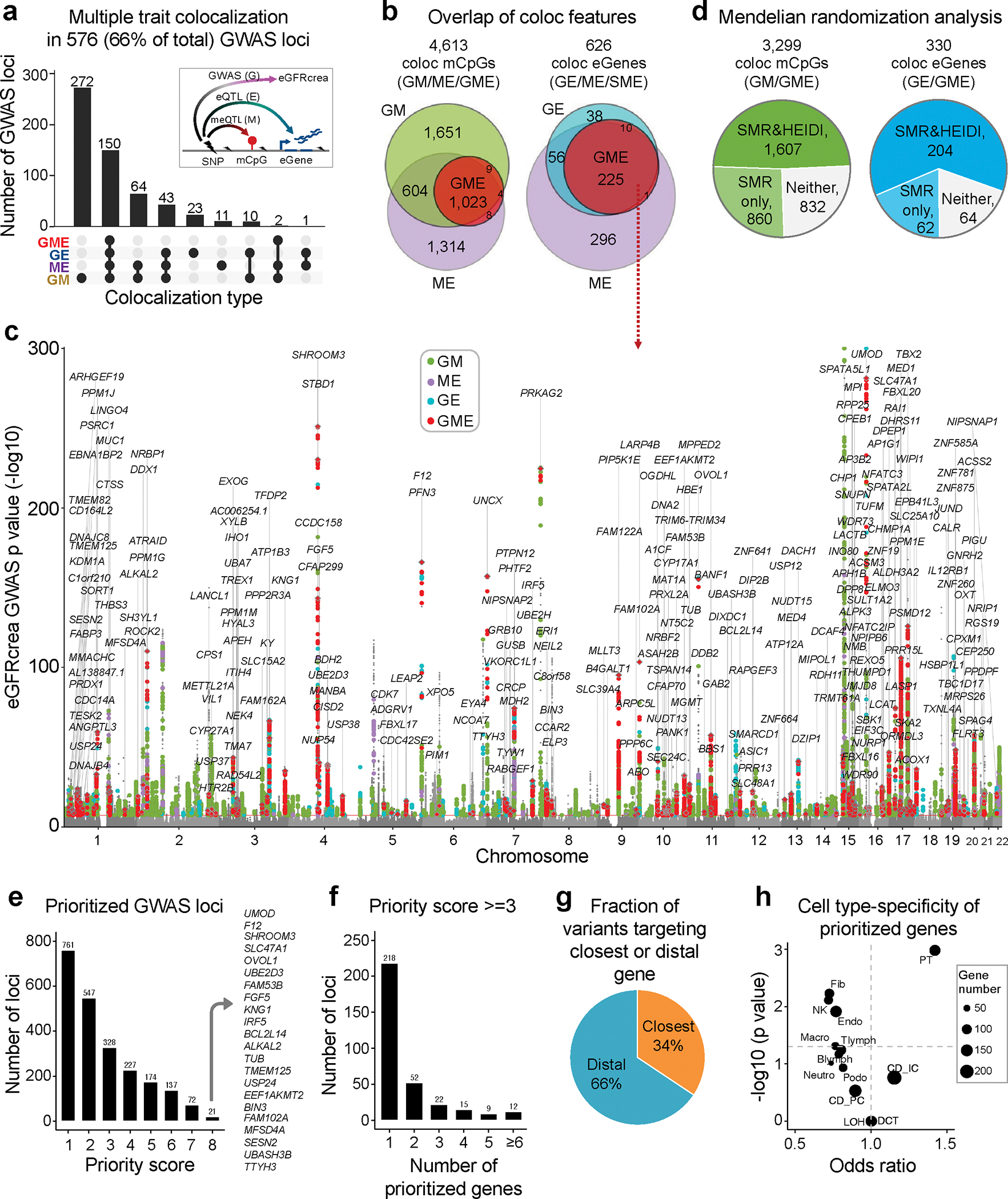
More than 800 million people suffer from kidney disease, yet the mechanism of kidney dysfunction is poorly understood. In the present study, we define the genetic association with kidney function in 1.5 million individuals and identify 878 (126 new) loci. We map the genotype effect on the methylome in 443 kidneys, transcriptome in 686 samples and single-cell open chromatin in 57,229 kidney cells. Heritability analysis reveals that methylation variation explains a larger fraction of heritability than gene expression. We present a multi-stage prioritization strategy and prioritize target genes for 87% of kidney function loci. We highlight key roles of proximal tubules and metabolism in kidney function regulation. Furthermore, the causal role of SLC47A1 in kidney disease is defined in mice with genetic loss of Slc47a1 and in human individuals carrying loss-of-function variants. Our findings emphasize the key role of bulk and single-cell epigenomic information in translating genome-wide association studies into identifying causal genes, cellular origins and mechanisms of complex traits.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests
The laboratory of Dr. Susztak receives funding from GSK, Regeneron, Gilead, Merck, Boehringer Ingelheim, Bayer, Novartis Maze, Jnana, Ventus and Novo Nordisk. The funders had no influence on the data analysis. Dr. Susztak serves on the SAB of Jnana pharmaceuticals and receives equity. Dr. Ritchie serves on the SAB for Goldfinch Bio and Cipherome. The other authors declare no competing interests.
Figures
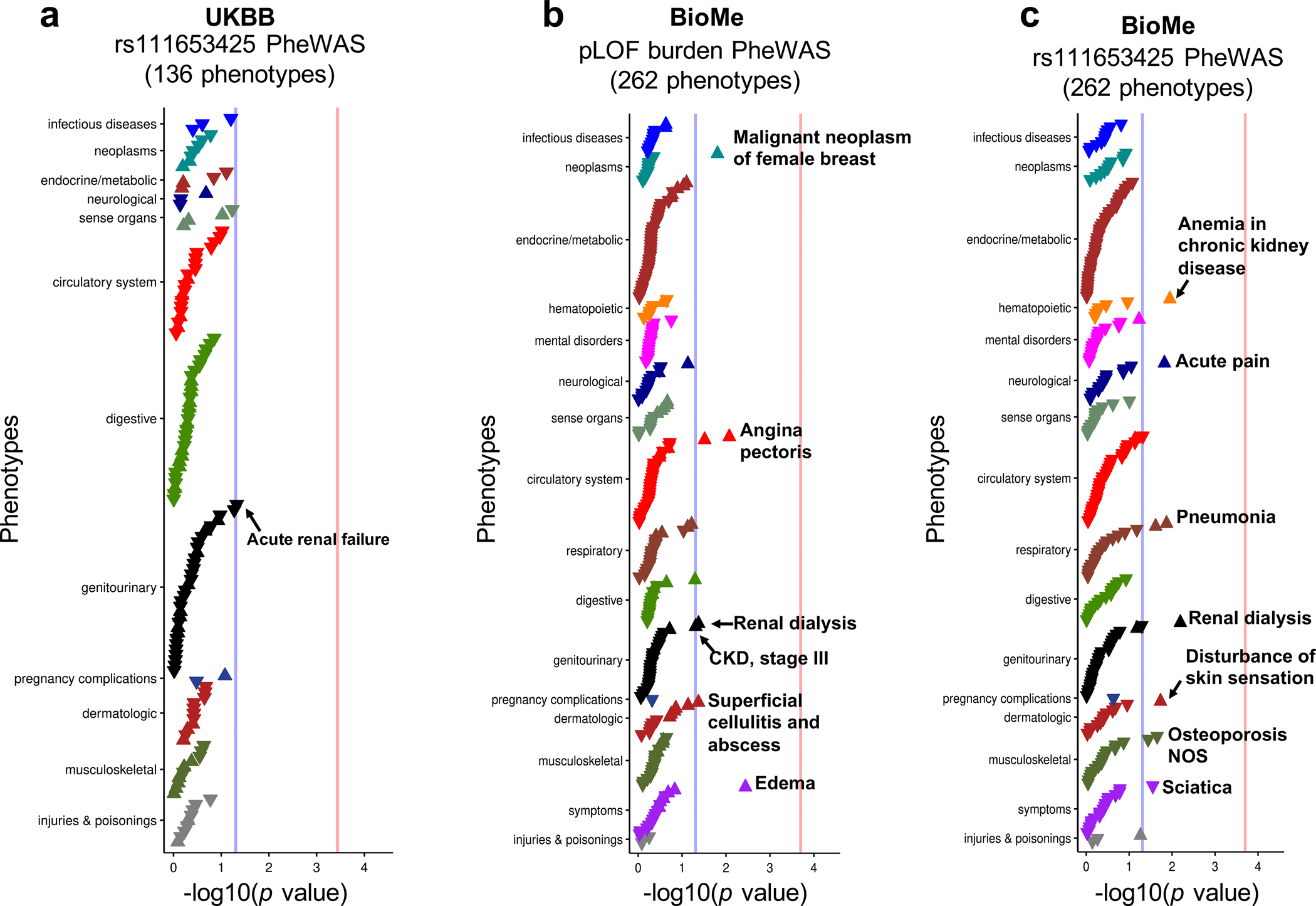
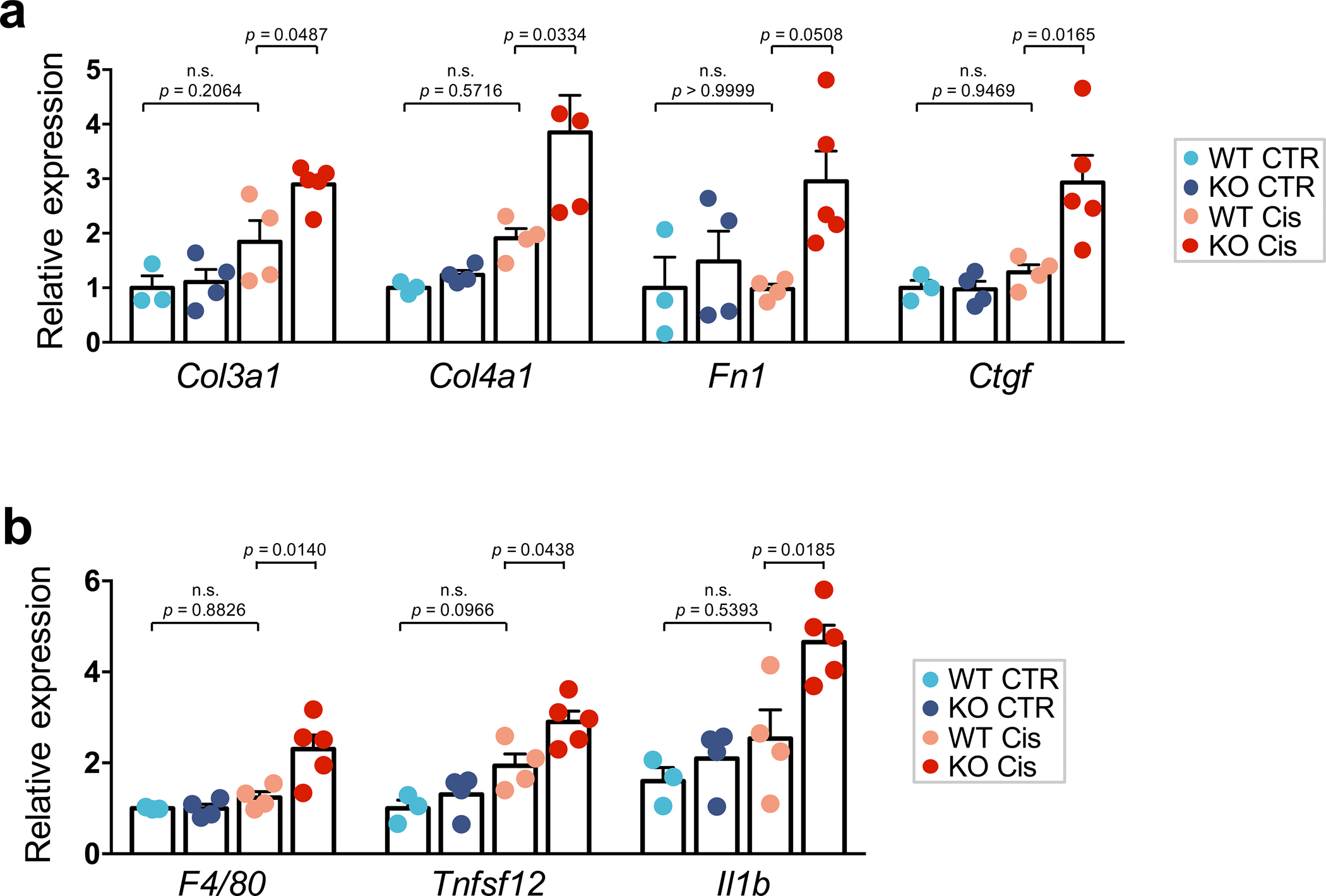
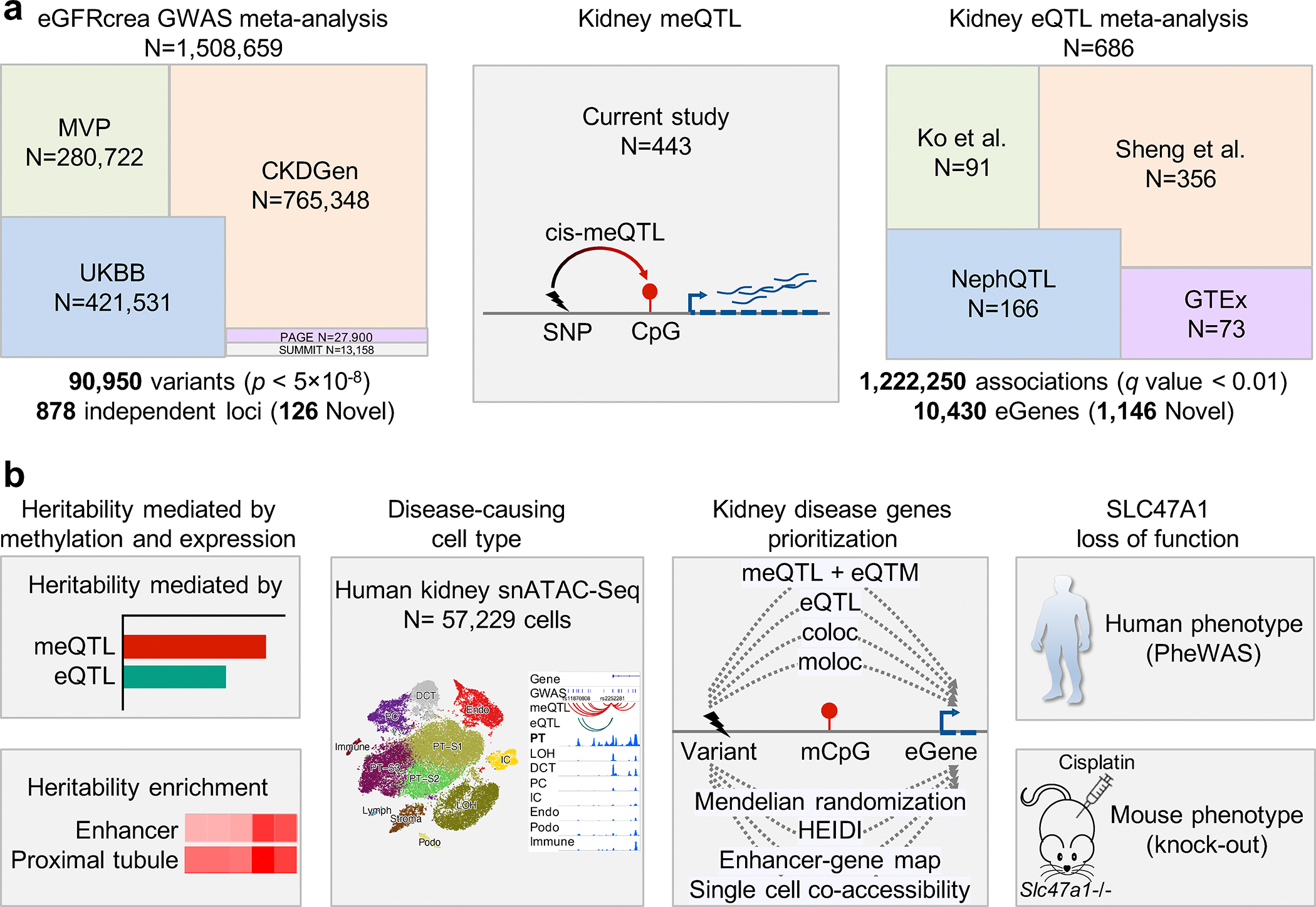
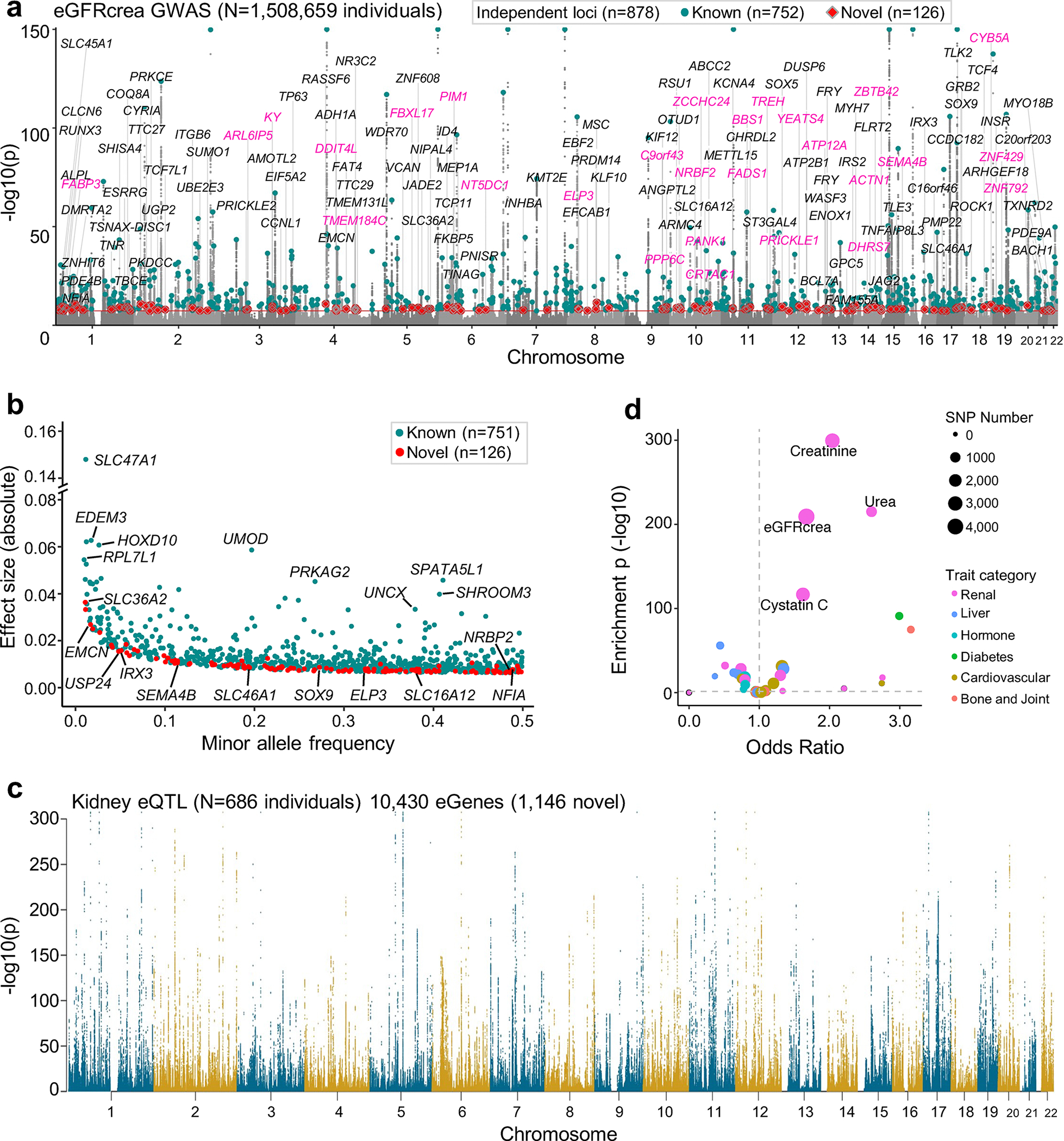
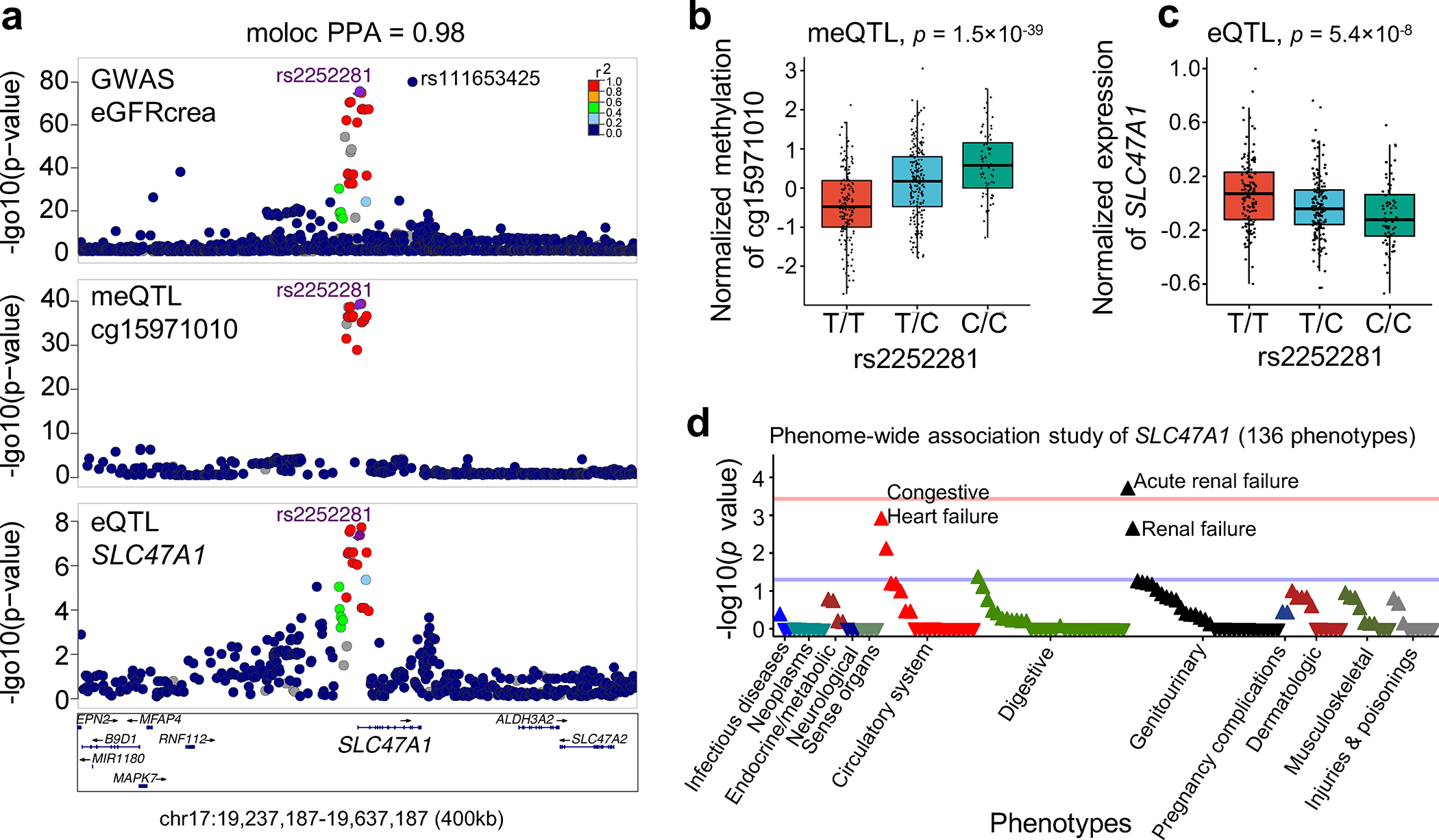
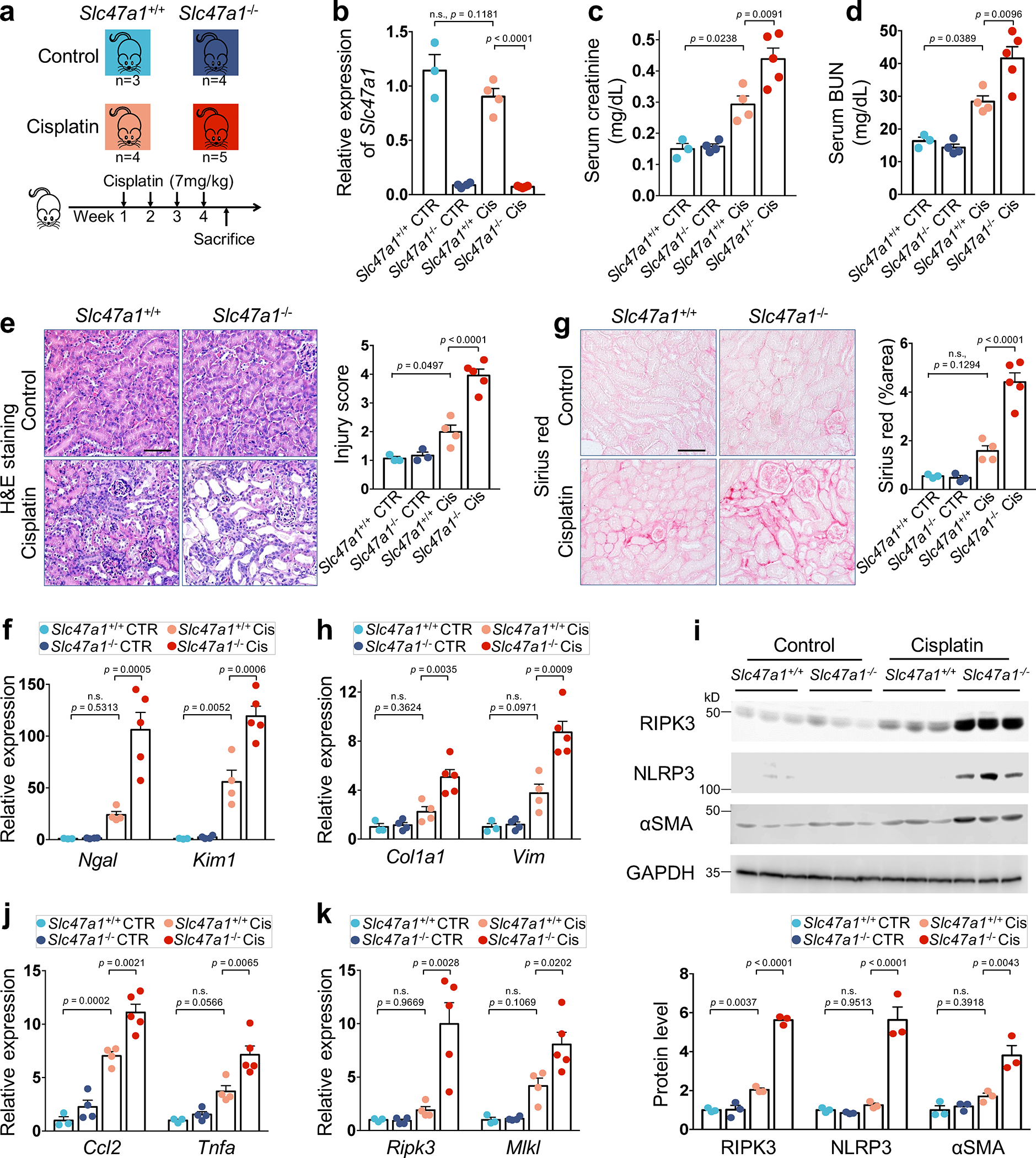
Comment in
-
On the way to translate GWAS into kidney disease mechanisms.Kidney Int. 2023 Jan;103(1):16-18. doi: 10.1016/j.kint.2022.10.004. Epub 2022 Oct 26. Kidney Int. 2023. PMID: 36309125 No abstract available.
References
Methods-only references
-
- Delaneau O, Zagury J-F & Marchini J Improved whole-chromosome phasing for disease and population genetic studies. Nature methods 10, 5 (2013). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
