

Mechanism of lesion verification by the human XPD helicase in nucleotide excision repair
- PMID: 35713557
- PMCID: PMC9262607
- DOI: 10.1093/nar/gkac496
Mechanism of lesion verification by the human XPD helicase in nucleotide excision repair
Abstract
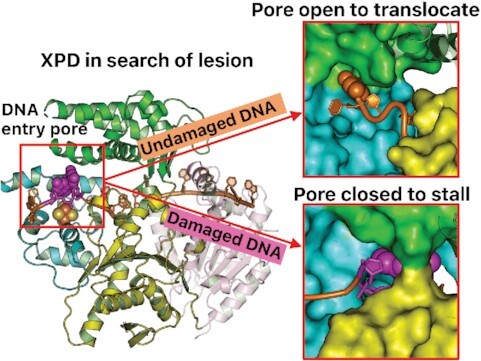
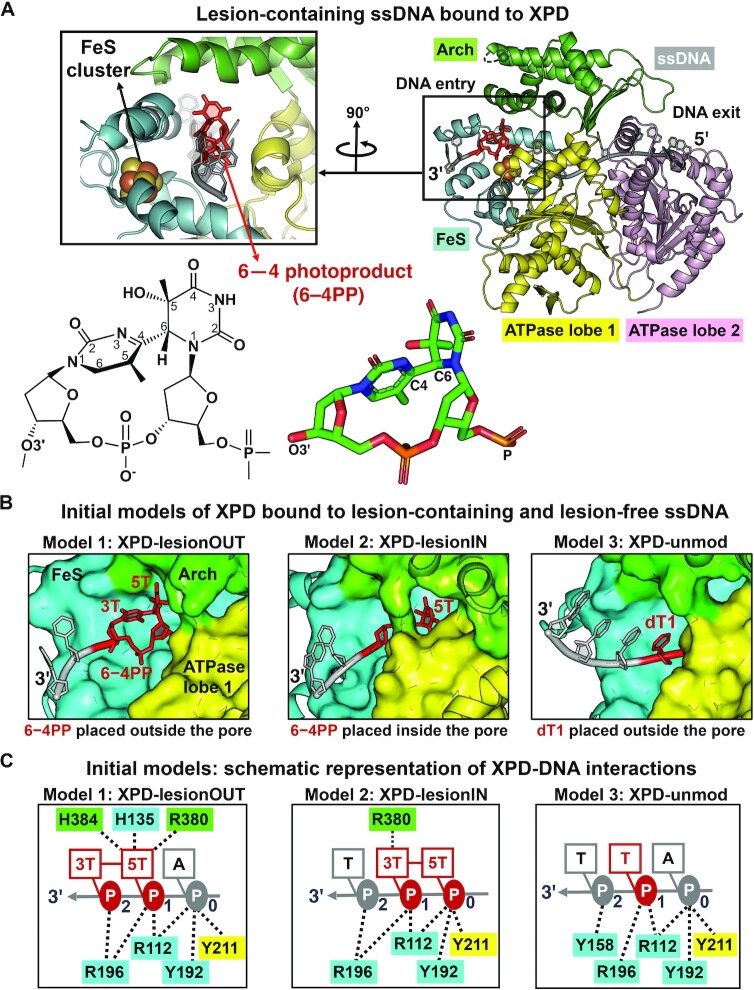
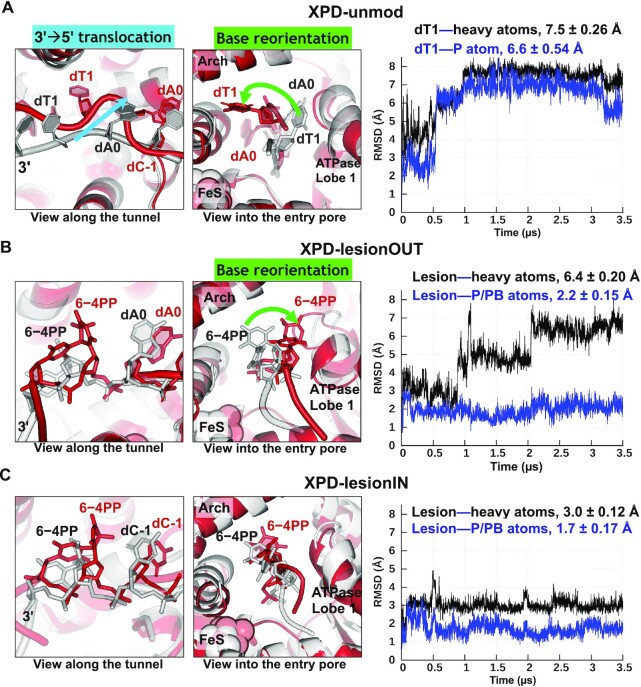
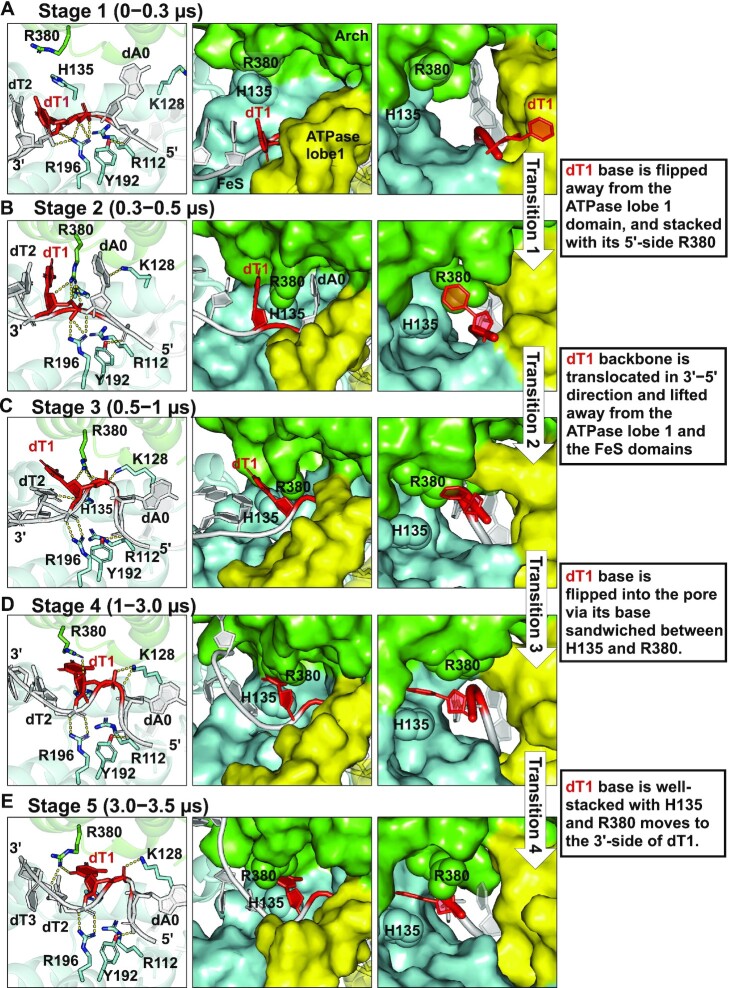
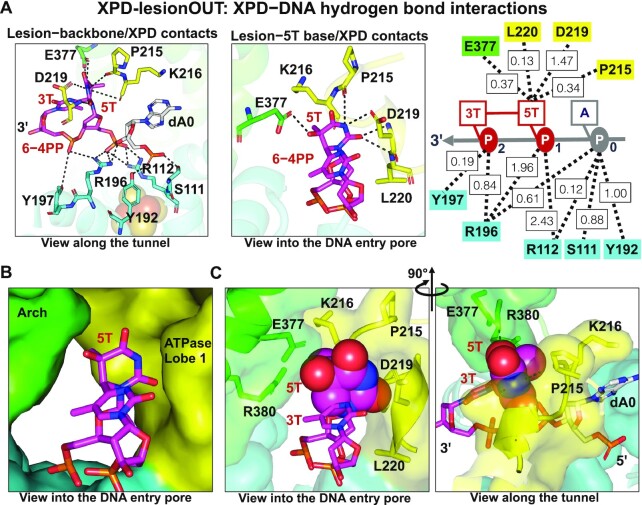
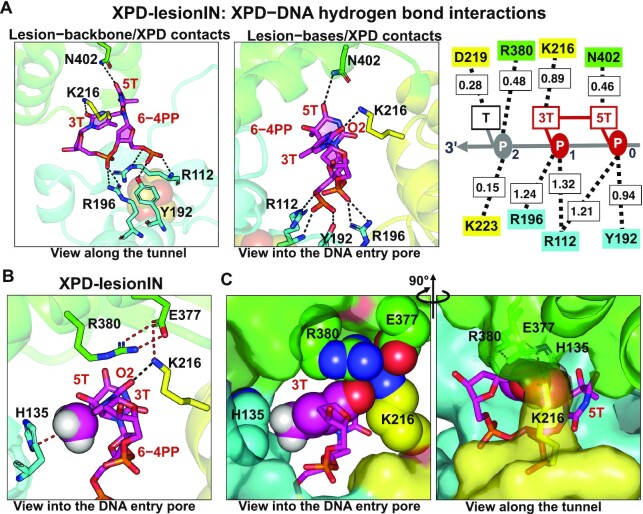
In nucleotide excision repair (NER), the xeroderma pigmentosum D helicase (XPD) scans DNA searching for bulky lesions, stalls when encountering such damage to verify its presence, and allows repair to proceed. Structural studies have shown XPD bound to its single-stranded DNA substrate, but molecular and dynamic characterization of how XPD translocates on undamaged DNA and how it stalls to verify lesions remains poorly understood. Here, we have performed extensive all-atom MD simulations of human XPD bound to undamaged and damaged ssDNA, containing a mutagenic pyrimidine (6-4) pyrimidone UV photoproduct (6-4PP), near the XPD pore entrance. We characterize how XPD responds to the presence of the DNA lesion, delineating the atomistic-scale mechanism that it utilizes to discriminate between damaged and undamaged nucleotides. We identify key amino acid residues, including FeS residues R112, R196, H135, K128, Arch residues E377 and R380, and ATPase lobe 1 residues 215-221, that are involved in damage verification and show how movements of Arch and ATPase lobe 1 domains relative to the FeS domain modulate these interactions. These structural and dynamic molecular depictions of XPD helicase activity with unmodified DNA and its inhibition by the lesion elucidate how the lesion is verified by inducing XPD stalling.
© The Author(s) 2022. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
