

Cryo-electron Microscopic Analysis of Single-Pass Transmembrane Receptors
- PMID: 35715229
- PMCID: PMC10026182
- DOI: 10.1021/acs.chemrev.1c01035
Cryo-electron Microscopic Analysis of Single-Pass Transmembrane Receptors
Abstract
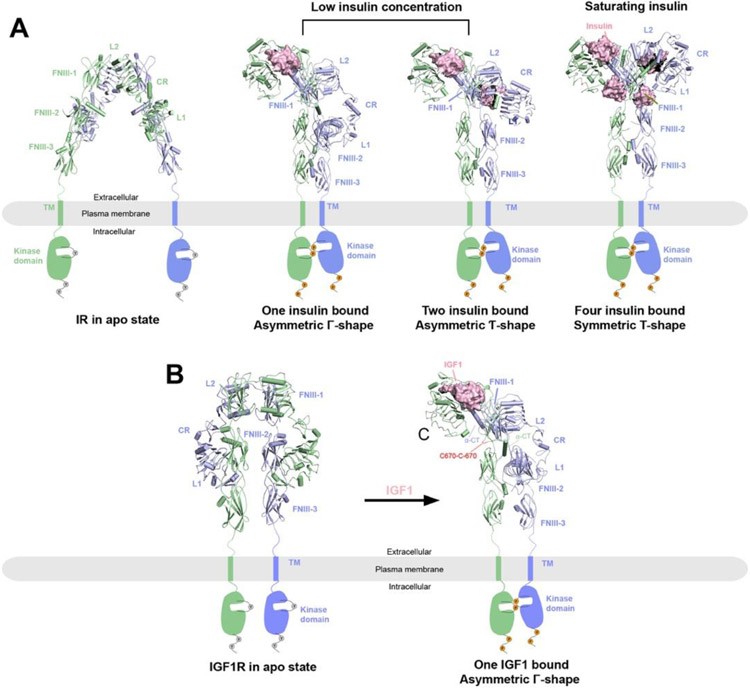
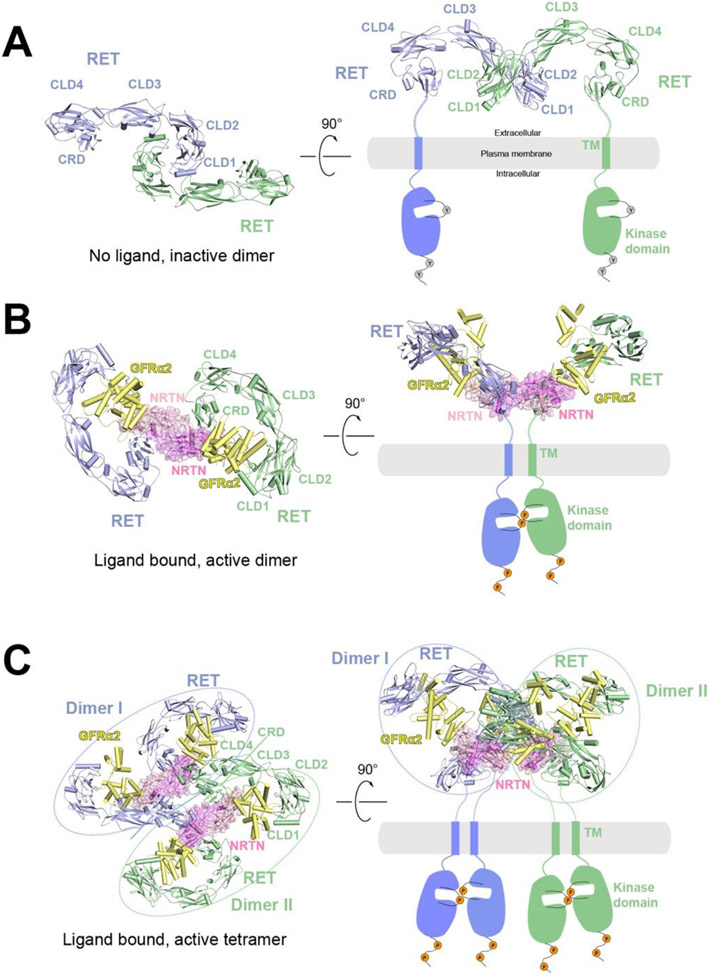
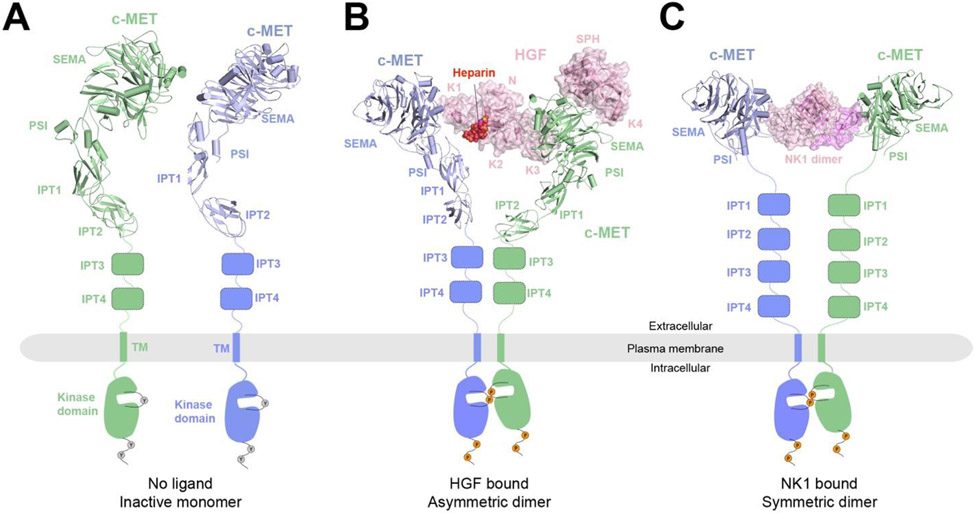
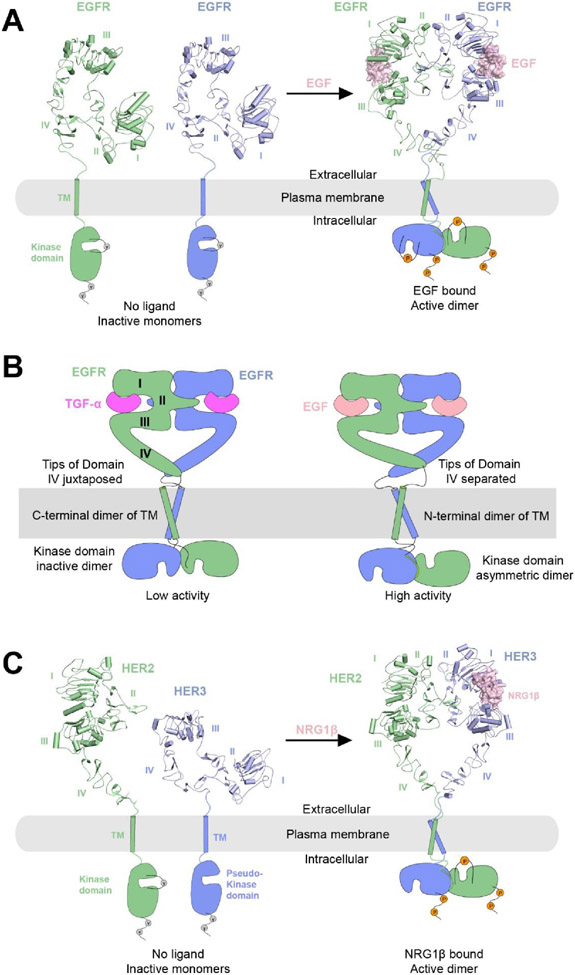
Single-pass transmembrane receptors (SPTMRs) represent a diverse group of integral membrane proteins that are involved in many essential cellular processes, including signal transduction, cell adhesion, and transmembrane transport of materials. Dysregulation of the SPTMRs is linked with many human diseases. Despite extensive efforts in past decades, the mechanisms of action of the SPTMRs remain incompletely understood. One major hurdle is the lack of structures of the full-length SPTMRs in different functional states. Such structural information is difficult to obtain by traditional structural biology methods such as X-ray crystallography and nuclear magnetic resonance (NMR). The recent rapid development of single-particle cryo-electron microscopy (cryo-EM) has led to an exponential surge in the number of high-resolution structures of integral membrane proteins, including SPTMRs. Cryo-EM structures of SPTMRs solved in the past few years have tremendously improved our understanding of how SPTMRs function. In this review, we will highlight these progresses in the structural studies of SPTMRs by single-particle cryo-EM, analyze important structural details of each protein involved, and discuss their implications on the underlying mechanisms. Finally, we also briefly discuss remaining challenges and exciting opportunities in the field.
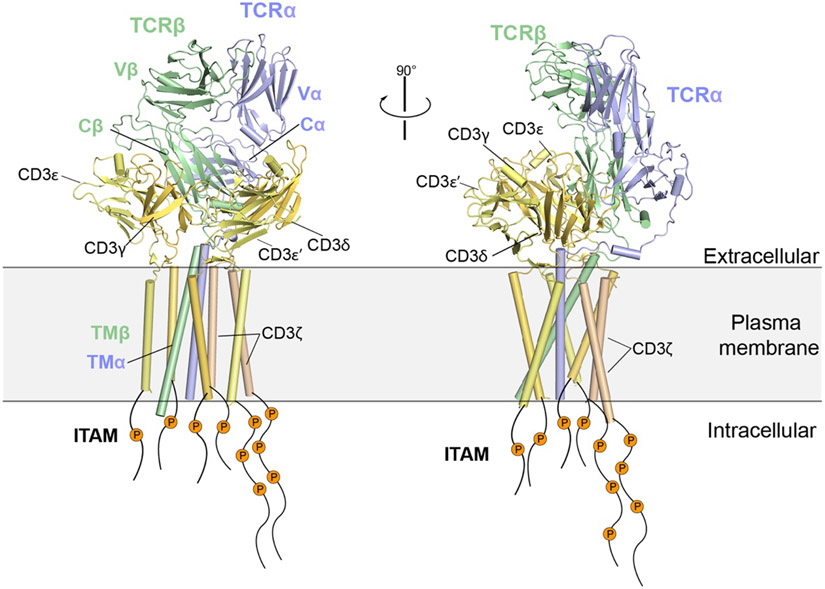
Figures
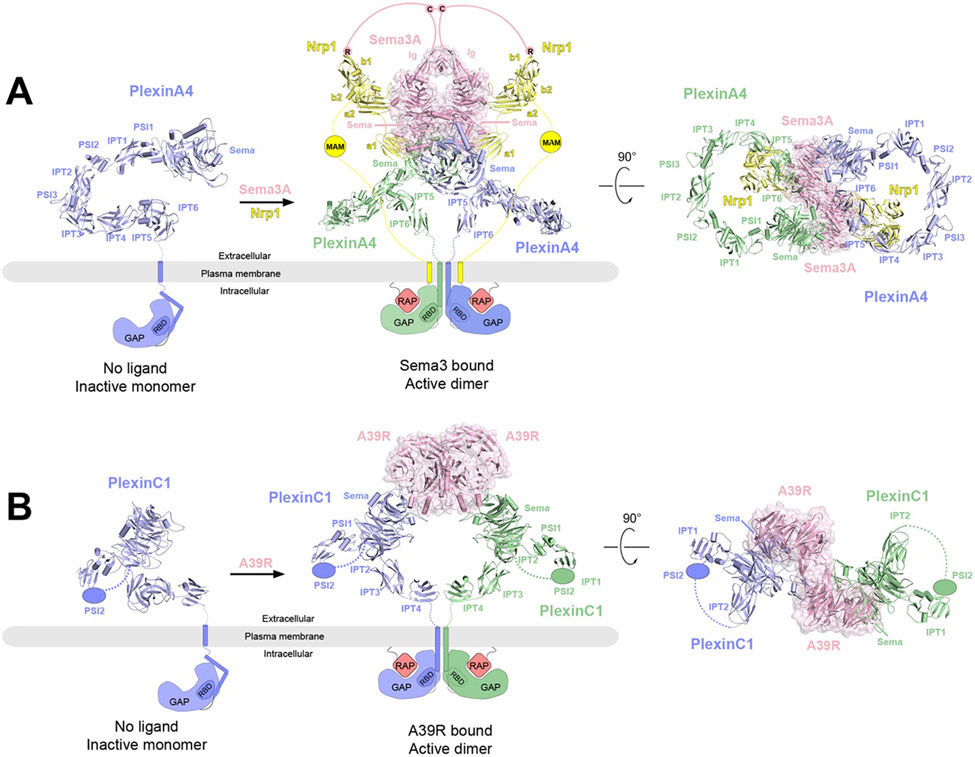
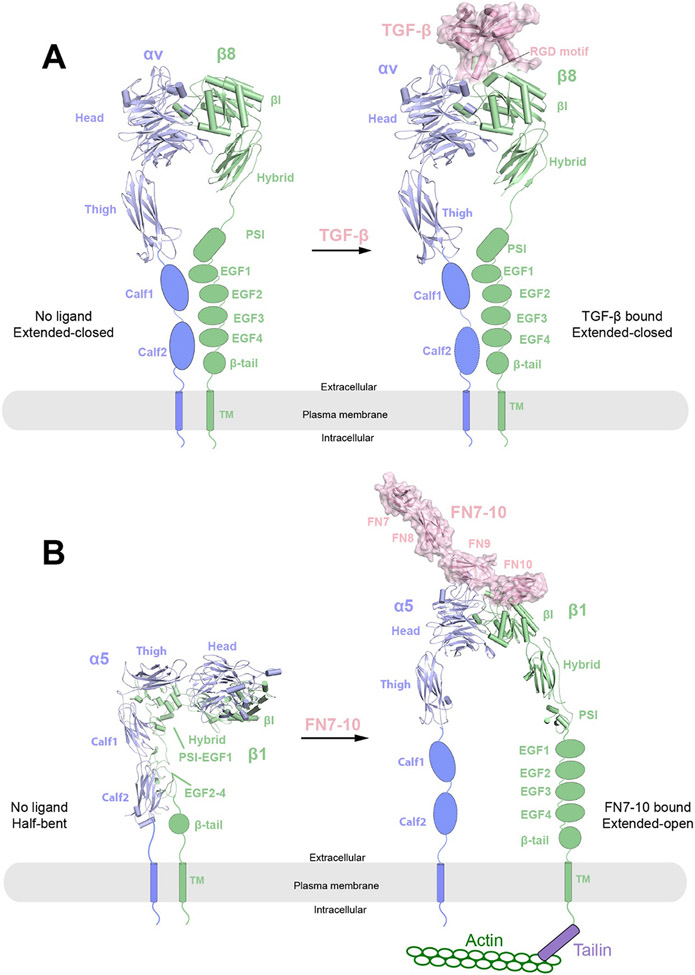
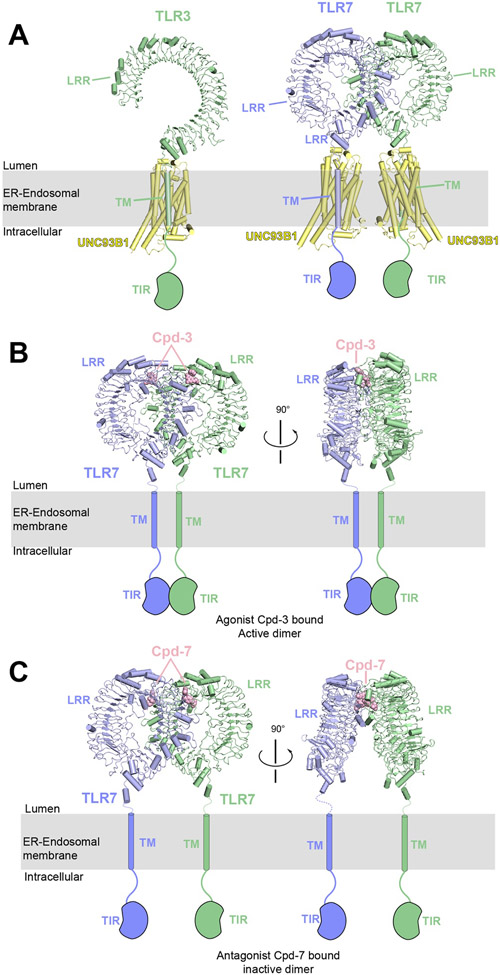
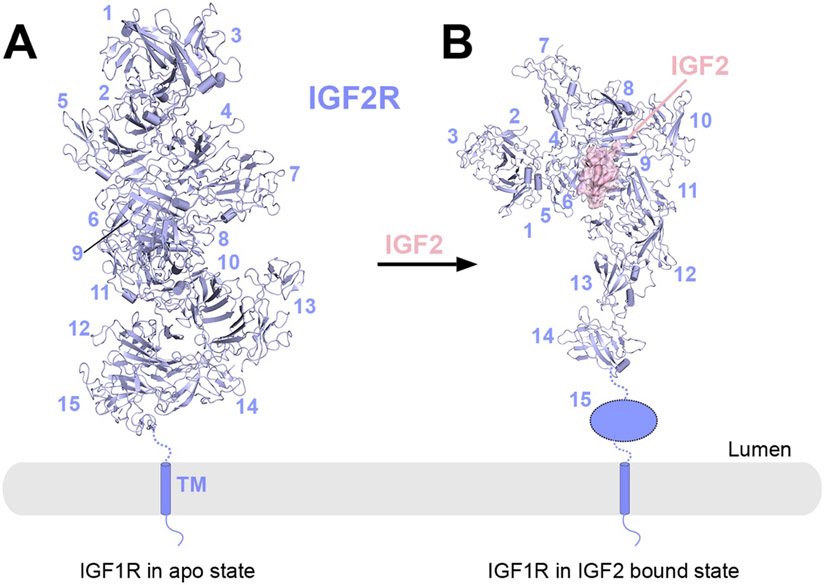
References
-
- Engel A; Gaub HE Structure and mechanics of membrane proteins. Annu. Rev. Biochem 2008, 77, 127–148. - PubMed
-
- Overington JP; Al-Lazikani B; Hopkins AL How many drug targets are there? Nat. Rev. Drug Discov 2006, 5, 993–996. - PubMed
-
- Bugge K; Lindorff-Larsen K; Kragelund BB Understanding single-pass transmembrane receptor signaling from a structural viewpoint-what are we missing? FEBS J. 2016, 283, 4424–4451. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
