

Onasemnogene abeparvovec for presymptomatic infants with three copies of SMN2 at risk for spinal muscular atrophy: the Phase III SPR1NT trial
- PMID: 35715567
- PMCID: PMC9205287
- DOI: 10.1038/s41591-022-01867-3
Onasemnogene abeparvovec for presymptomatic infants with three copies of SMN2 at risk for spinal muscular atrophy: the Phase III SPR1NT trial
Abstract
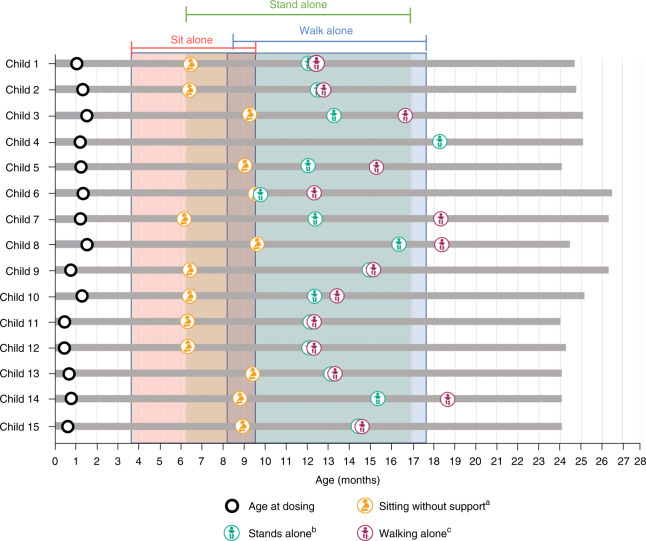
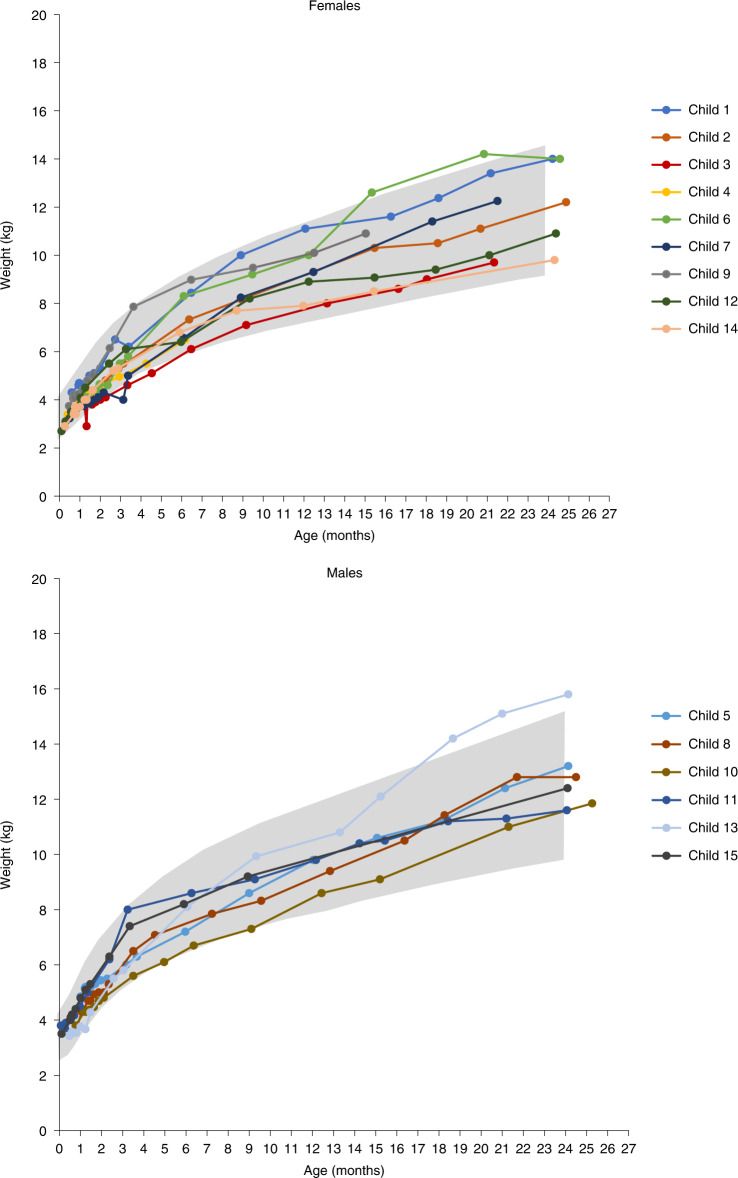
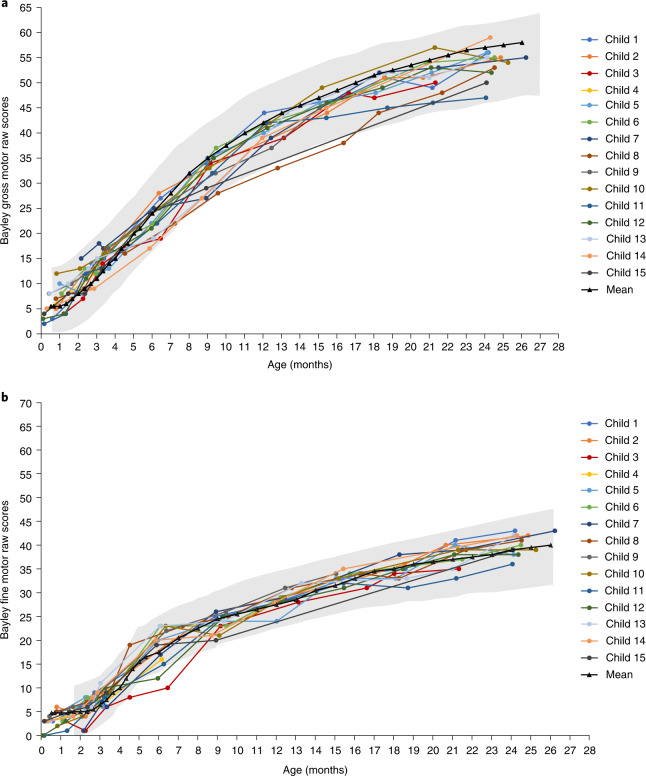
Most children with biallelic SMN1 deletions and three SMN2 copies develop spinal muscular atrophy (SMA) type 2. SPR1NT ( NCT03505099 ), a Phase III, multicenter, single-arm trial, investigated the efficacy and safety of onasemnogene abeparvovec for presymptomatic children with biallelic SMN1 mutations treated within six postnatal weeks. Of 15 children with three SMN2 copies treated before symptom onset, all stood independently before 24 months (P < 0.0001; 14 within normal developmental window), and 14 walked independently (P < 0.0001; 11 within normal developmental window). All survived without permanent ventilation at 14 months; ten (67%) maintained body weight (≥3rd WHO percentile) without feeding support through 24 months; and none required nutritional or respiratory support. No serious adverse events were considered treatment-related by the investigator. Onasemnogene abeparvovec was effective and well-tolerated for presymptomatic infants at risk of SMA type 2, underscoring the urgency of early identification and intervention.
© 2022. The Author(s).
Conflict of interest statement
Novartis Gene Therapies, Inc. sponsored this clinical trial. The authors declare the following competing interests. K.A.S. has received personal compensation from Novartis Gene Therapies, Inc. (formerly AveXis, Inc.) for serving as an advisory board member and from Biogen for serving as a visiting professor, and has received research support from clinical trials sponsored by Novartis Gene Therapies, Inc., and Biogen. M.A.F. has received honoraria for scientific advisory boards from Novartis Gene Therapies, Inc., Biogen, and Roche and research grants from Biogen. F.M. reports grants and personal fees from Novartis Gene Therapies, Inc., including participation in the STR1VE-EU grant and the SPR1NT study; grants, personal fees, and other (participation in the SHINE clinical trial and principal investigator of the investigator-initiated UK SMA REACH UK registry) from Biogen; and grants and personal fees from Roche, including participation in the JEWELFISH clinical trial of risdiplam and participation in the olesoxime clinical trial, during the conduct of the study. F.M. has also received honoraria for scientific advisory boards from Biogen, Novartis, Novartis Gene Therapies, Inc., PTC, Roche, and Sarepta Therapeutics. K.S. has served on advisory boards for Biogen, Novartis Gene Therapies, Inc./Novartis, and Chugai (Roche) and has received search funding from Biogen. J.R.M. has received personal compensation for clinical trial consulting and for serving on scientific advisory boards, as well as research support, from Novartis Gene Therapies, Inc. L.S. has received personal compensation as an advisory committee board member/consultant from Novartis Gene Therapies, Inc., Biogen, Biophytis, Cytokinetics, Dynacure, Roche, Santhera, and Sarepta Therapeutics, and has received research support from Novartis Gene Therapies, Inc., Biogen, Dynacure, and Roche. H.J.M. has received honoraria for scientific advisory boards from Novartis Gene Therapies, Inc., and has received research funding from Roche. R.S.F. has received personal compensation for advisory board participation from Novartis Gene Therapies, Inc., Biogen, Roche, and Scholar Rock, and for consulting from Novartis; editorial fees from Elsevier for co-editing a neurology textbook; license fees from the Children’s Hospital of Philadelphia; and research funding from Novartis Gene Therapies, Inc., Biogen, Roche/Genentech, and Scholar Rock. K.J.S. has received personal compensation for a speaking engagement from Biogen, and research funding as a principal investigator for clinical trials sponsored by Novartis Gene Therapies, Inc. and Biogen. J.M.K. was site principal investigator for clinical trials sponsored by Novartis Gene Therapies, Inc., Biogen, and Scholar Rock. C.M.Z. has received research support from Biogen. C.A.C. has served on advisory boards for Novartis Gene Therapies, Inc., and Roche/Genentech; has served as an educational speaker for Biogen; and has received research funding from Novartis Gene Therapies, Inc., Biogen, and Roche. S.T.I. has received personal compensation for service on advisory boards or consulting from Novartis Gene Therapies, Inc., Biogen, Roche/Genentech, and Sarepta Therapeutics; and research support from Novartis Gene Therapies, Inc., Biogen, Capricor, PTC, Scholar Rock, and Sarepta Therapeutics. J.M.K. is a principal investigator for clinical trials sponsored by Novartis Gene Therapies, Inc., Scholar Rock, and Roche/Genentech. J.A.P. has served as an investigator on clinical trials for Biogen, Novartis, PTC Therapeutics, and Scholar Rock, and has served in an advisory capacity for Biogen, Scholar Rock, Roche/Genentech, and Novartis. P.B.S. has served as a consultant for Alexion, Biogen, Genentech, Novartis Gene Therapies, Inc., and Sarepta and has served as a speaker for Alexion, Biogen, Genentech, Grifols, Novartis Gene Therapies, Inc., and PTC Therapeutics. S.K. is an employee of Novartis Gene Therapies, Inc., and receives consulting fees from UCB Pharma, Karuna Therapeutics, Worldwide Clinical Trials, CPC Clinical Research, Zosano Pharmaceuticals, PharPoint Research, and Nesos, Inc. M.W. is an employee of Novartis Gene Therapies, Inc., and owns Novartis stock or other equities. S.T.-W. is an employee of Novartis Gene Therapies, Inc., and owns Novartis stock or other equities. B.E.M. is an employee of Translational Medicine, Novartis Institutes for BioMedical Research (Cambridge), and owns Novartis stock or other equities. T.A.M. is an employee of Novartis Gene Therapies, Inc., and owns Novartis stock or other equities.
Figures
Comment in
-
Early treatment is a lifeline for infants with SMA.Nat Med. 2022 Jul;28(7):1348-1349. doi: 10.1038/s41591-022-01889-x. Nat Med. 2022. PMID: 35840728 Free PMC article.
-
Timing is everything: Clinical evidence supports pre-symptomatic treatment for spinal muscular atrophy.Cell Rep Med. 2022 Aug 16;3(8):100725. doi: 10.1016/j.xcrm.2022.100725. Cell Rep Med. 2022. PMID: 35977471 Free PMC article.
References
-
- Coovert DD, et al. The survival motor neuron protein in spinal muscular atrophy. Hum. Mol. Genet. 1997;6:1205–1214. - PubMed
-
- Mailman MD, et al. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet. Med. 2002;4:20–26. - PubMed
-
- Calucho M, et al. Correlation between SMA type and SMN2 copy number revisited: an analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul. Disord. 2018;28:208–215. - PubMed
-
- Carson VJ, et al. Nusinersen by subcutaneous intrathecal catheter for symptomatic spinal muscular atrophy patients with complex spine anatomy. Muscle Nerve. 2022;65:51–59. - PubMed
-
- Muntoni F, et al. Long-term follow-up of patients with type 2 and non-ambulant type 3 spinal muscular atrophy (SMA) treated with olesoxime in the OLEOS trial. Neuromuscul. Disord. 2020;30:959–969. - PubMed
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
