

LILRB2-mediated TREM2 signaling inhibition suppresses microglia functions
- PMID: 35717259
- PMCID: PMC9206387
- DOI: 10.1186/s13024-022-00550-y
LILRB2-mediated TREM2 signaling inhibition suppresses microglia functions
Abstract
Background: Microglia plays crucial roles in Alzheimer's disease (AD) development. Triggering receptor expressed on myeloid cells 2 (TREM2) in association with DAP12 mediates signaling affecting microglia function. Here we study the negative regulation of TREM2 functions by leukocyte immunoglobulin-like receptor subfamily B member 2 (LILRB2), an inhibitory receptor bearing ITIM motifs.
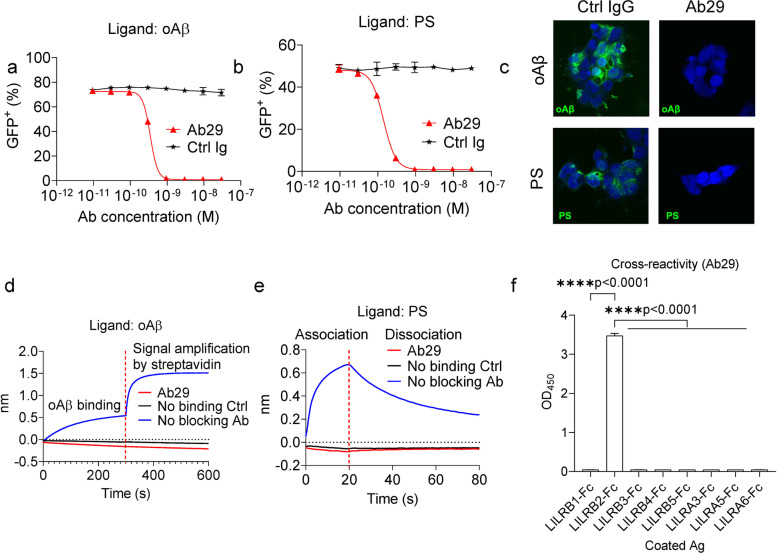
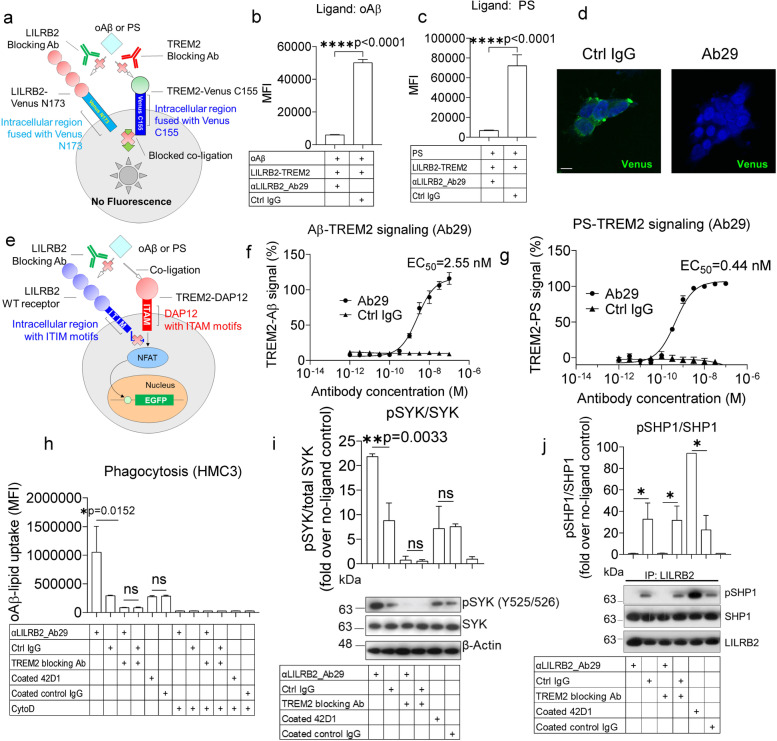
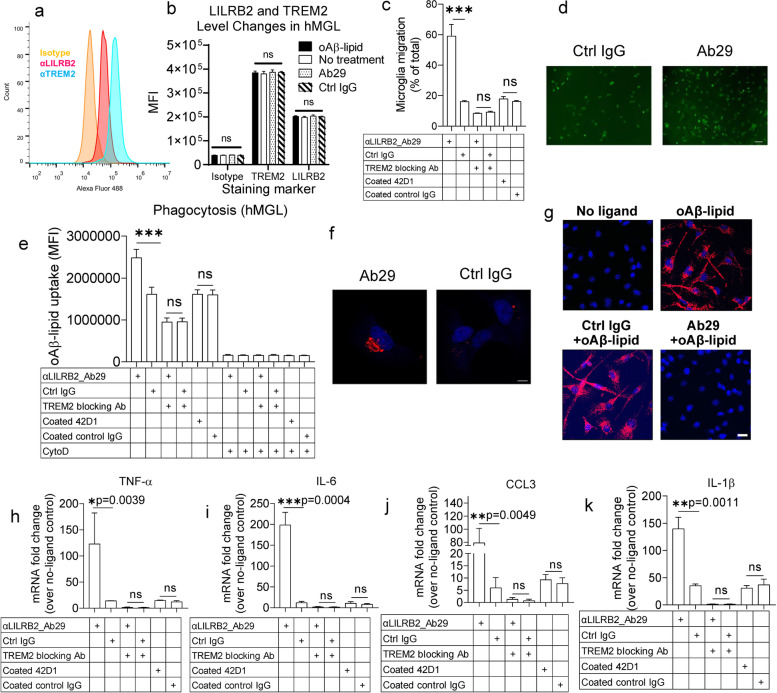
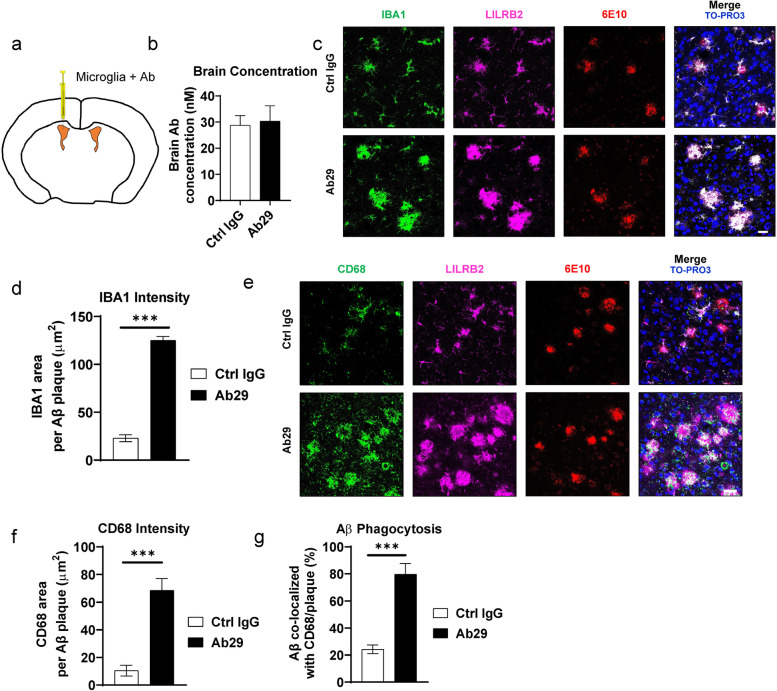
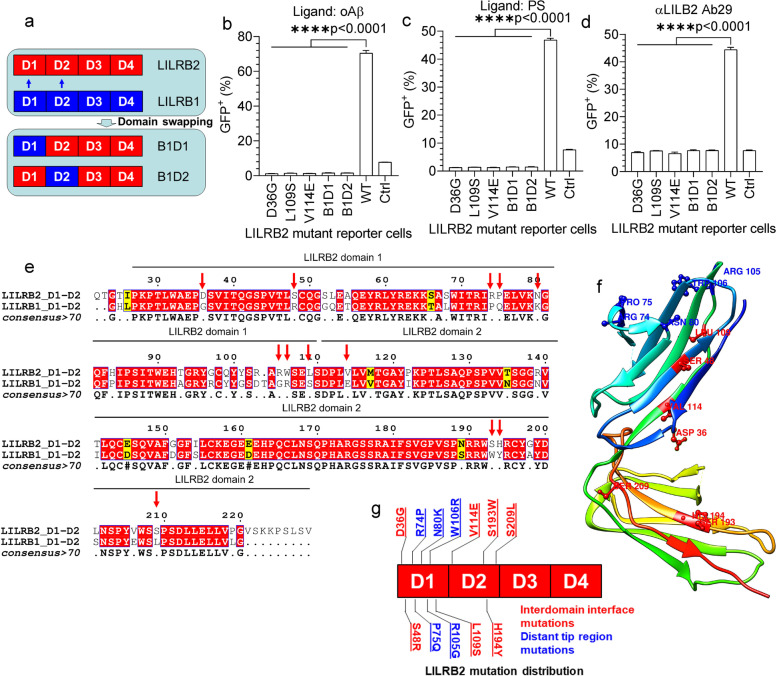
Methods: To specifically interrogate LILRB2-ligand (oAβ and PS) interactions and microglia functions, we generated potent antagonistic LILRB2 antibodies with sub-nanomolar level activities. The biological effects of LILRB2 antagonist antibody (Ab29) were studied in human induced pluripotent stem cell (iPSC)-derived microglia (hMGLs) for migration, oAβ phagocytosis, and upregulation of inflammatory cytokines. Effects of the LILRB2 antagonist antibody on microglial responses to amyloid plaques were further studied in vivo using stereotaxic grafted microglia in 5XFAD mice.
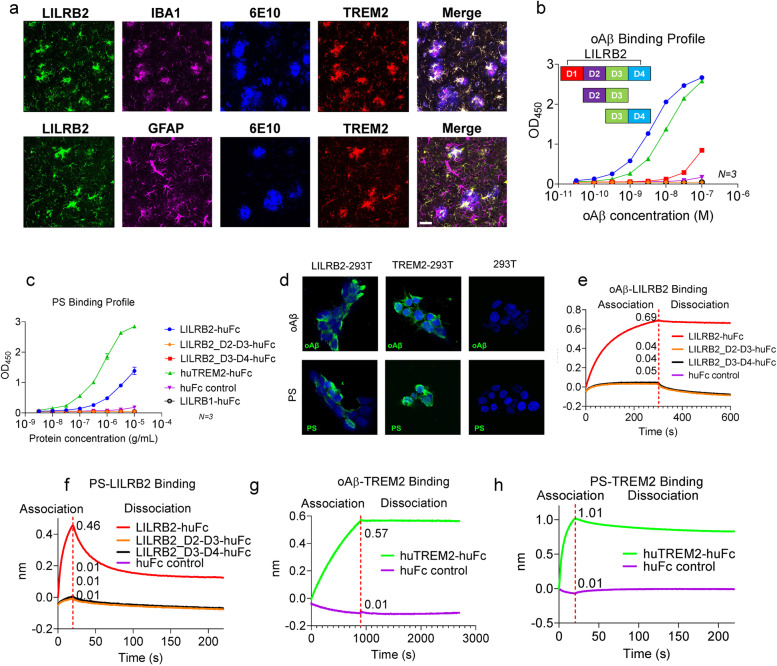
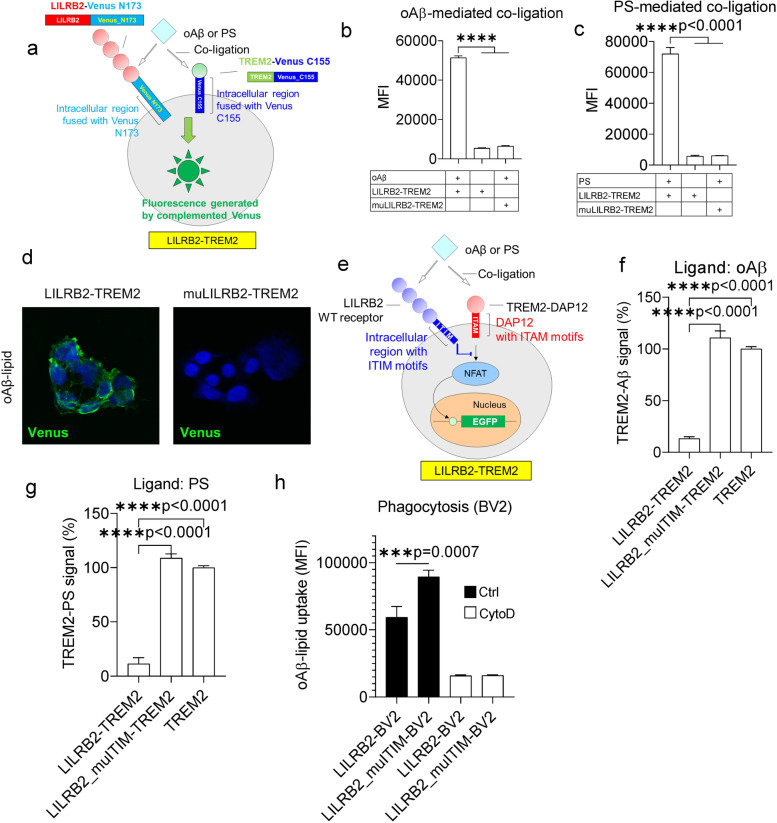
Results: We confirmed the expression of both LILRB2 and TREM2 in human brain microglia using immunofluorescence. Upon co-ligation of the LILRB2 and TREM2 by shared ligands oAβ or PS, TREM2 signaling was significantly inhibited. We identified a monoclonal antibody (Ab29) that blocks LILRB2/ligand interactions and prevents TREM2 signaling inhibition mediated by LILRB2. Further, Ab29 enhanced microglia phagocytosis, TREM2 signaling, migration, and cytokine responses to the oAβ-lipoprotein complex in hMGL and microglia cell line HMC3. In vivo studies showed significantly enhanced clustering of microglia around plaques with a prominent increase in microglial amyloid plaque phagocytosis when 5XFAD mice were treated with Ab29.
Conclusions: This study revealed for the first time the molecular mechanisms of LILRB2-mediated inhibition of TREM2 signaling in microglia and demonstrated a novel approach of enhancing TREM2-mediated microglia functions by blocking LILRB2-ligand interactions. Translationally, a LILRB2 antagonist antibody completely rescued the inhibition of TREM2 signaling by LILRB2, suggesting a novel therapeutic strategy for improving microglial functions.
Keywords: 5XFAD mice; Alzheimer’s disease; Amyloid; Antibody; ITAM; ITIM; LILRB2; Microglia; Phagocytosis; TREM2.
© 2022. The Author(s).
Conflict of interest statement
The University of Texas System has filed a patent application on the LILRB2 targeting antibodies and PZ, NZ, and ZA are named inventors of the patent application.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
