

Clinical and genetic profile of patients enrolled in the Transthyretin Amyloidosis Outcomes Survey (THAOS): 14-year update
- PMID: 35717381
- PMCID: PMC9206752
- DOI: 10.1186/s13023-022-02359-w
Clinical and genetic profile of patients enrolled in the Transthyretin Amyloidosis Outcomes Survey (THAOS): 14-year update
Abstract
Background: Transthyretin amyloidosis (ATTR amyloidosis) is a rare, life-threatening disease caused by the accumulation of variant or wild-type (ATTRwt amyloidosis) transthyretin amyloid fibrils in the heart, peripheral nerves, and other tissues and organs.
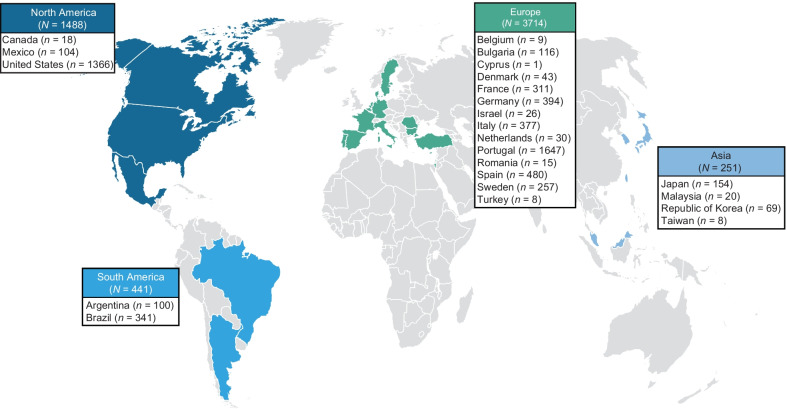
Methods: Established in 2007, the Transthyretin Amyloidosis Outcomes Survey (THAOS) is the largest ongoing, global, longitudinal observational study of patients with ATTR amyloidosis, including both inherited and wild-type disease, and asymptomatic carriers of pathogenic TTR mutations. This descriptive analysis examines baseline characteristics of symptomatic patients and asymptomatic gene carriers enrolled in THAOS since its inception in 2007 (data cutoff: August 1, 2021).
Results: This analysis included 3779 symptomatic patients and 1830 asymptomatic gene carriers. Symptomatic patients were predominantly male (71.4%) and had a mean (standard deviation [SD]) age of symptom onset of 56.3 (17.8) years. Val30Met was the most common genotype in symptomatic patients in South America (80.9%), Europe (55.4%), and Asia (50.5%), and more patients had early- versus late-onset disease in these regions. The majority of symptomatic patients in North America (58.8%) had ATTRwt amyloidosis. The overall distribution of phenotypes in symptomatic patients was predominantly cardiac (40.7%), predominantly neurologic (40.1%), mixed (16.6%), and no phenotype (2.5%). In asymptomatic gene carriers, mean (SD) age at enrollment was 42.4 (15.7) years, 42.4% were male, and 73.2% carried the Val30Met mutation.
Conclusions: This 14-year global overview of THAOS in over 5000 patients represents the largest analysis of ATTR amyloidosis to date and highlights the genotypic and phenotypic heterogeneity of the disease.
Clinicaltrials: gov Identifier: NCT00628745.
Keywords: Amyloidosis; Cardiomyopathy; Polyneuropathy; Registry; Transthyretin.
© 2022. The Author(s).
Conflict of interest statement
AD reports research grants from Celgene, Millennium, Pfizer, Janssen, and Alnylam; and has received funding from Pfizer for meeting expenses (travel) and attended advisory boards for Akcea and Intellia. TC reports serving as a medical advisor for Pfizer and receiving funding for scientific meeting expenses (travel, accommodation, and registration) from Pfizer. IC reports consulting fees and funding for scientific meeting expenses (travel, accommodation, and registration) from Pfizer. MW-C reports research funding, consulting fees, and travel support for advisory boards and meetings from FoldRx Pharmaceuticals and Pfizer. JW reports consulting fees and travel support for lectures and advisory boards from Pfizer and Alnylam, and consulting fees from Akcea. AVK reports reimbursement for study visits from Pfizer during the conduct of the study. CR reports research grants from Pfizer and consultancy fees from Pfizer, Alnylam, and Prothena. VP-B reports serving as a medical advisor for Pfizer, Alnylam, Eidos, and Ionis. JG-M reports speaker fees from Pfizer, Alnylam, and Akcea. MSM reports grants from Pfizer during the conduct of the study; grants and personal fees from Pfizer, Eidos, Prothena, and Ionis; grants from Alnylam; and personal fees from GSK and Akcea outside the submitted work. MG reports grants and advisory board and consultancy fees paid to her institution from Alnylam, Eidos, Prothena, and Pfizer. DC and LA are full-time employees of Pfizer and hold stock and/or stock options with Pfizer.
Figures




References
-
- Rowczenio D, Wechalekar A. Mutations in hereditary amyloidosis. 2015. http://amyloidosismutations.com/mut-attr.php. Accessed 11 Oct 2021.
Publication types
MeSH terms
Substances
Supplementary concepts
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
