

Functional Properties of Two Distinct PTH1R Mutants Associated With Either Skeletal Defects or Pseudohypoparathyroidism
- PMID: 35720667
- PMCID: PMC9189904
- DOI: 10.1002/jbm4.10604
Functional Properties of Two Distinct PTH1R Mutants Associated With Either Skeletal Defects or Pseudohypoparathyroidism
Abstract
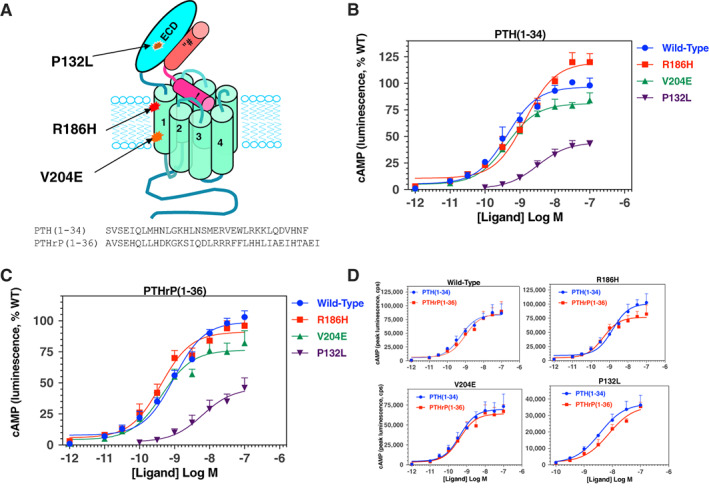
Consistent with a vital role of parathyroid hormone (PTH) receptor type 1 (PTH1R) in skeletal development, homozygous loss-of-function PTH1R mutations in humans results in neonatal lethality (Blomstrand chondrodysplasia), whereas such heterozygous mutations cause a primary failure of tooth eruption (PFE). Despite a key role of PTH1R in calcium and phosphate homeostasis, blood mineral ion levels are not altered in such cases of PFE. Recently, two nonlethal homozygous PTH1R mutations were identified in two unrelated families in which affected members exhibit either dental and skeletal abnormalities (PTH1R-V204E) or hypocalcemia and hyperphosphatemia (PTH1R-R186H). Arg186 and Val204 map to the first transmembrane helix of the PTH1R, and thus to a critical region of this class B G protein-coupled receptor. We used cell-based assays and PTH and PTH-related protein (PTHrP) ligand analogs to assess the impact of the R186H and V204E mutations on PTH1R function in vitro. In transiently transfected HEK293 cells, PTH1R-R186H mediated cyclic adenosine monophosphate (cAMP) responses to PTH(1-34) and PTHrP(1-36) that were of comparable potency to those observed on wild-type PTH1R (PTH1R-WT) (half maximal effective concentrations [EC50s] = 0.4nM to 1.2nM), whereas the response-maxima were significantly reduced for the PTH1R-V204E mutant (maximum effect [Emax] = 81%-77% of PTH1R-WT, p ≤ 0.004). Antibody binding to an extracellular hemagglutinin (HA) tag was comparable for PTH1R-R186H and PTH1R-WT, but was significantly reduced for PTH1R-V204E (maximum binding level [Bmax] = 44% ± 11% of PTH1R-WT, p = 0.002). The potency of cAMP signaling induced by a PTH(1-11) analog was reduced by ninefold and threefold, respectively, for PTH1R-R186H and PTH1R-V204E, relative to PTH1R-WT, and a PTH(1-15) radioligand analog that bound adequately to PTH1R-WT exhibited little or no specific binding to either mutant receptor. The data support a general decrease in PTH1R surface expression and/or function as a mechanism for PFE and a selective impairment in PTH ligand affinity as a potential PTH1R-mutation-based mechanism for pseudohypoparathyroidism. © 2022 The Authors. JBMR Plus published by Wiley Periodicals LLC on behalf of American Society for Bone and Mineral Research.
Keywords: HYPERPHOSPHATEMIA; HYPOCALCEMIA; PARATHYROID; PRIMARY FAILURE OF TOOTH ERUPTION; PSEUDOHYPOPARATHYROIDISM; PTH; PTH1R; PTHrP.
© 2022 The Authors. JBMR Plus published by Wiley Periodicals LLC on behalf of American Society for Bone and Mineral Research.
Conflict of interest statement
The authors have no conflicts of interest for the generation of this manuscript.
Figures






References
-
- Duchatelet S, Ostergaard E, Cortes D, Lemainque A, Julier C. Recessive mutations in PTHR1 cause contrasting skeletal dysplasias in Eiken and Blomstrand syndromes. Hum Mol Genet. 2005;14:1‐5. - PubMed
-
- Hoogendam J, Farih‐Sips H, Wynaendts LC, Lowik CW, Wit JM, Karperien M. Novel mutations in the parathyroid hormone (PTH)/PTH‐related peptide receptor type 1 causing Blomstrand osteochondrodysplasia types I and II. J Clin Endocrinol Metab. 2007;92:1088‐1095. - PubMed
-
- Karaplis AC, He B, Nguyen MT, et al. Inactivating mutation in the human parathyroid hormone receptor type 1 gene in Blomstrand chondrodysplasia. Endocrinology. 1998;139:5255‐5258. - PubMed
-
- Oostra RJ, van der Harten JJ, Rijnders WP, Scott RJ, Young MP, Trump D. Blomstrand osteochondrodysplasia: three novel cases and histological evidence for heterogeneity. Virchows Arch. 2000;436:28‐35. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials