

Single-cell analyses define a continuum of cell state and composition changes in the malignant transformation of polyps to colorectal cancer
- PMID: 35726067
- PMCID: PMC9279149
- DOI: 10.1038/s41588-022-01088-x
Single-cell analyses define a continuum of cell state and composition changes in the malignant transformation of polyps to colorectal cancer
Abstract
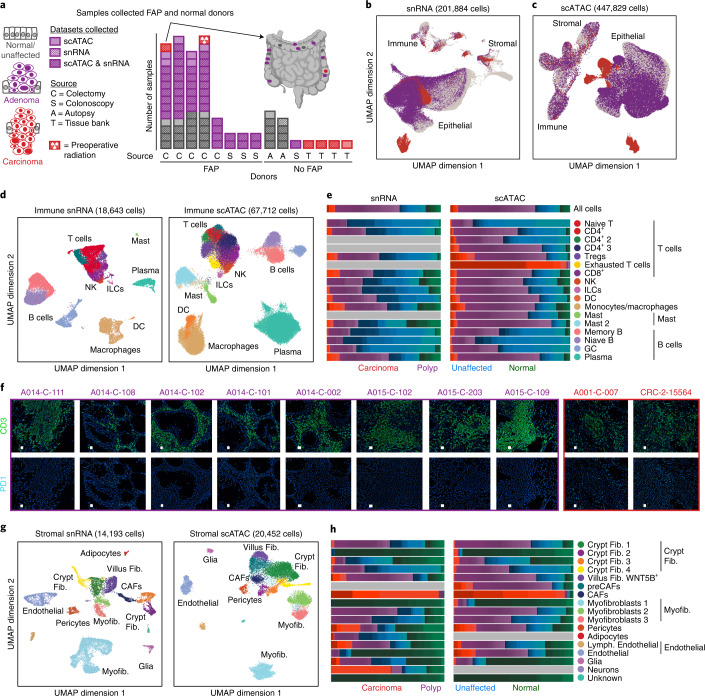
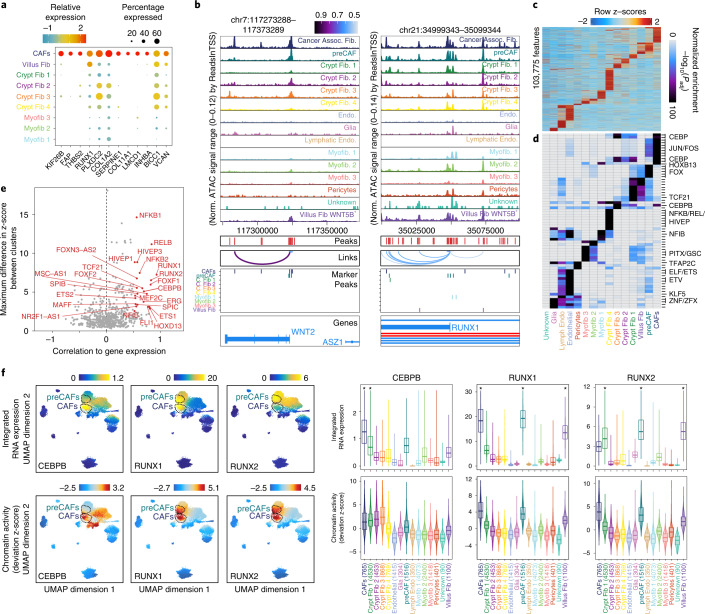
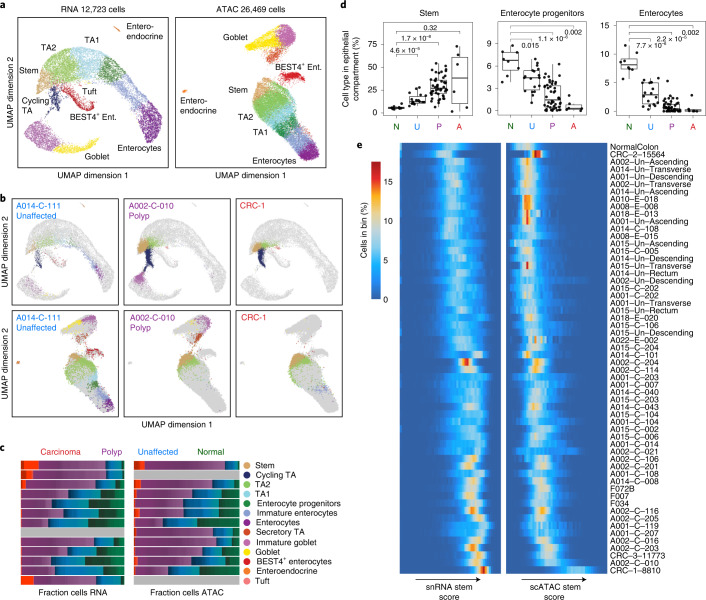
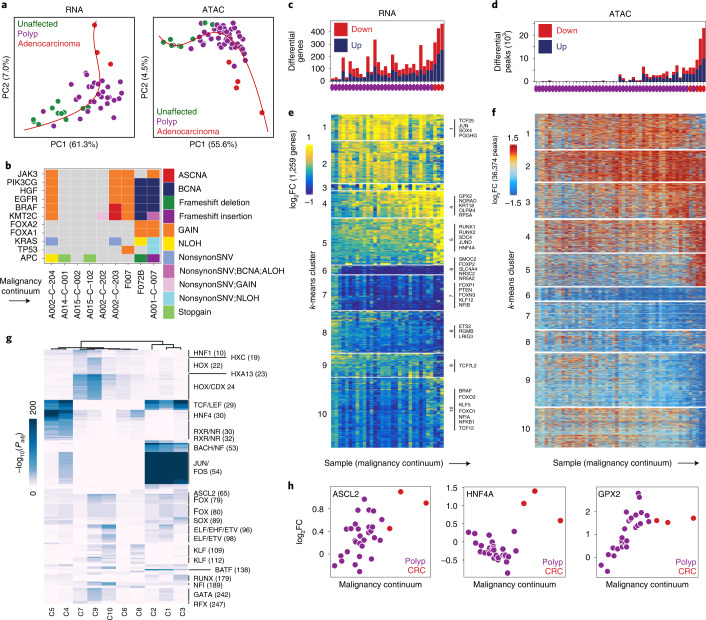
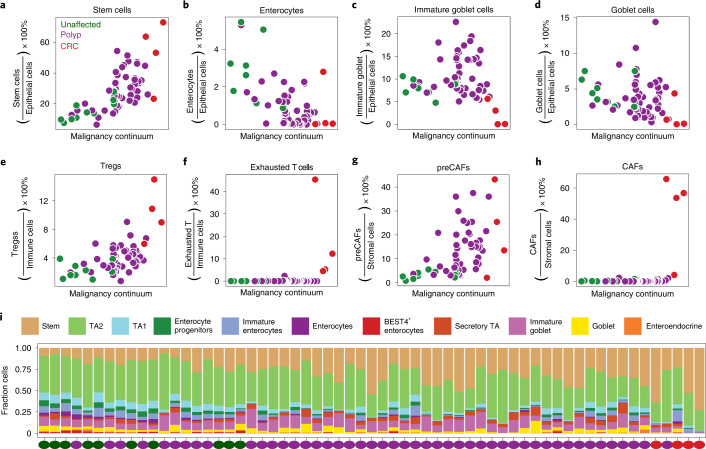
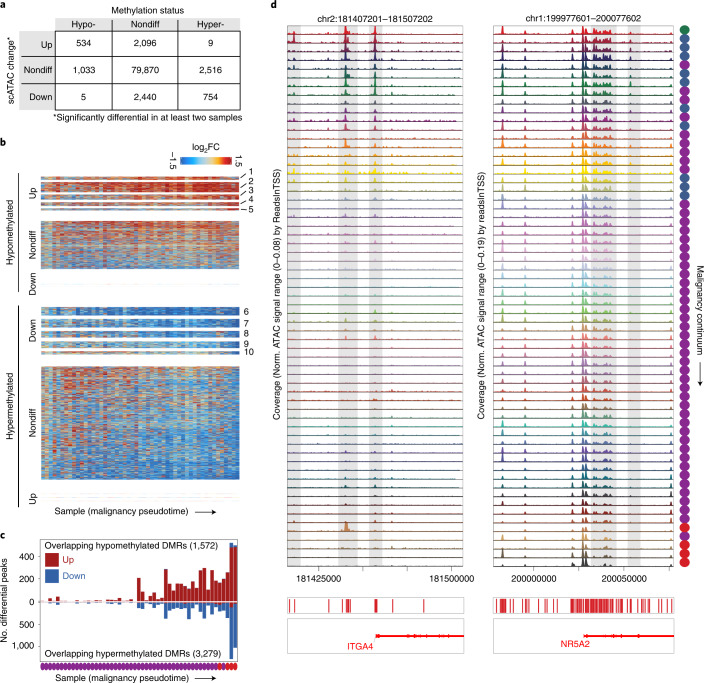
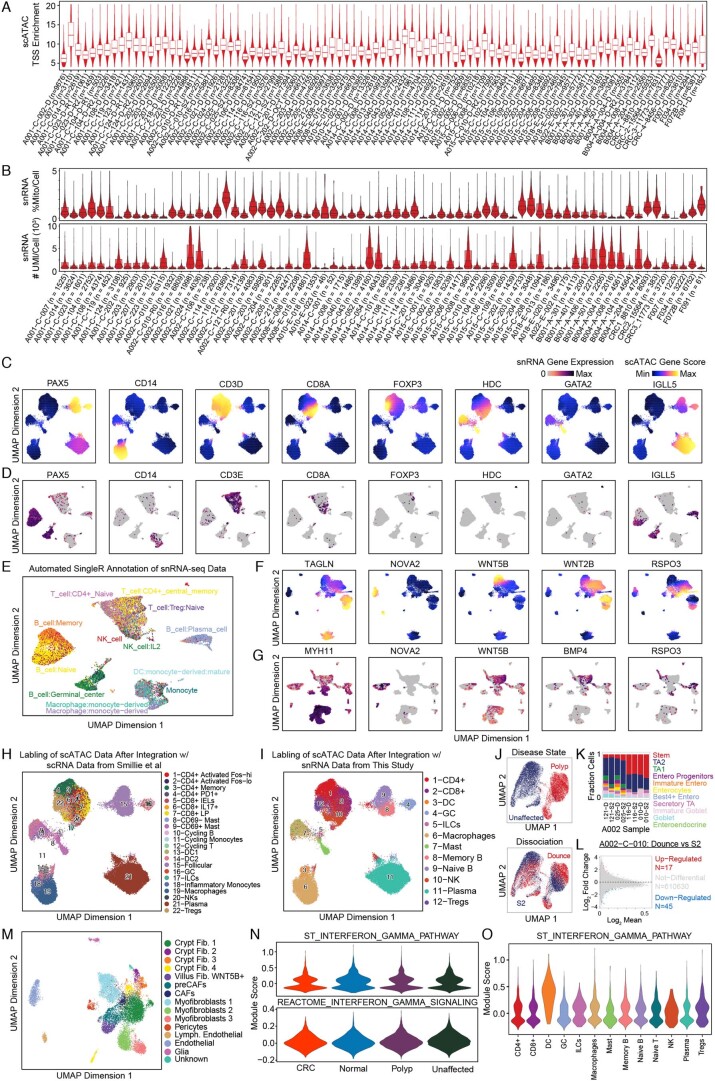
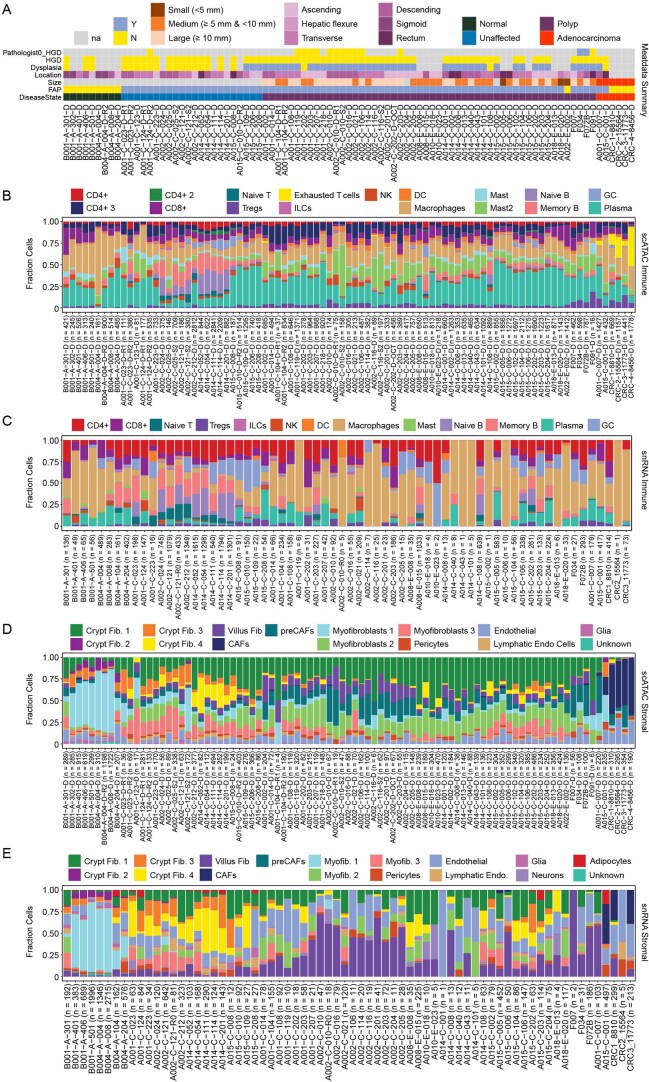
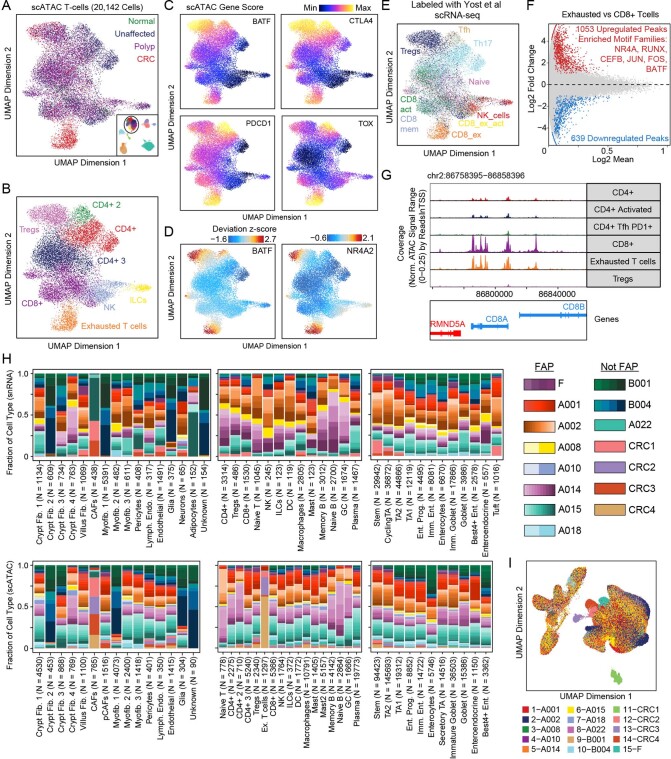
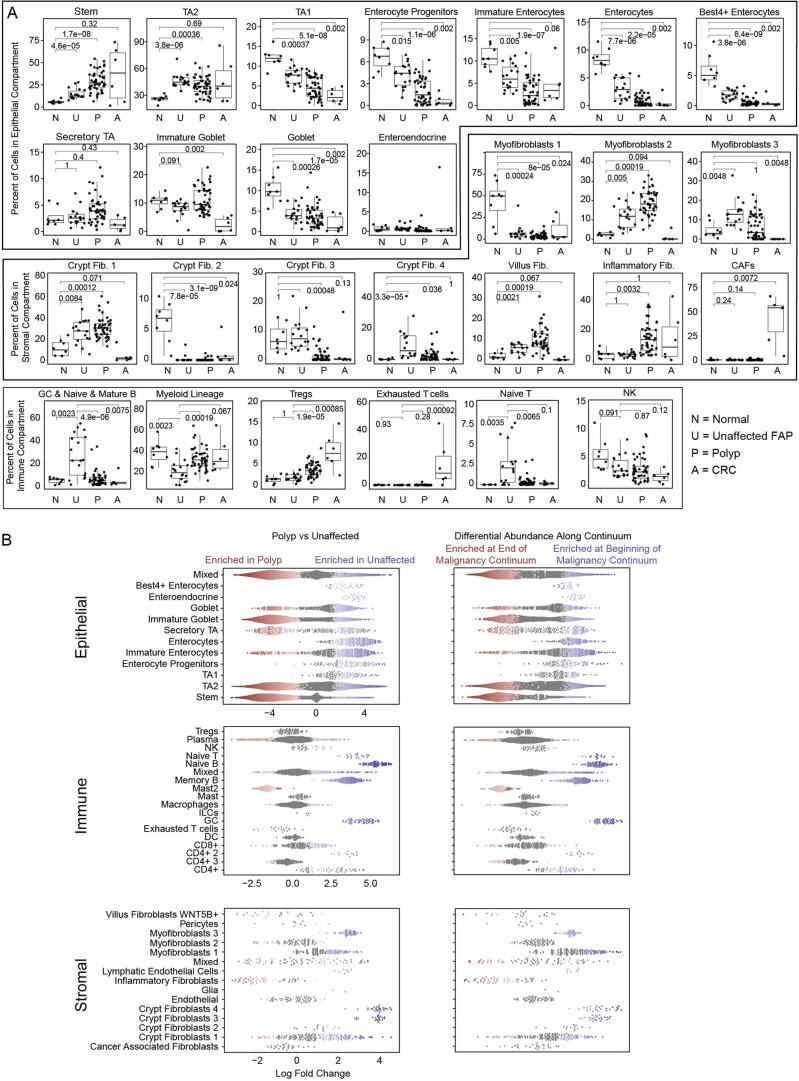
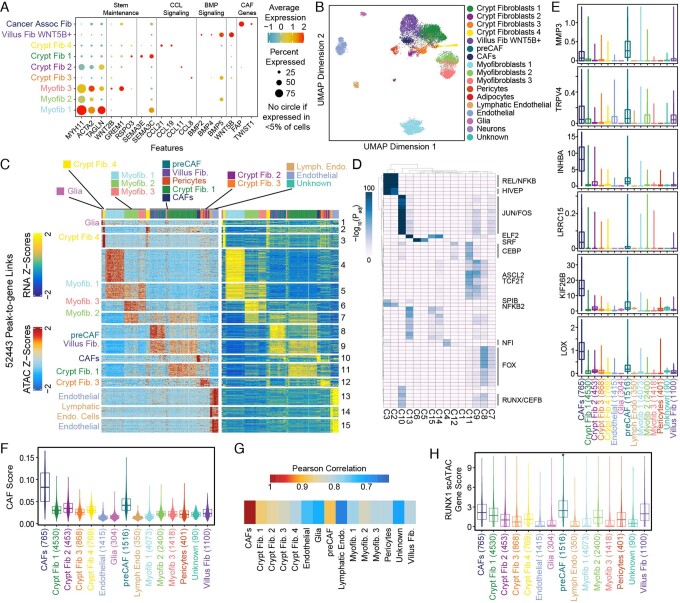
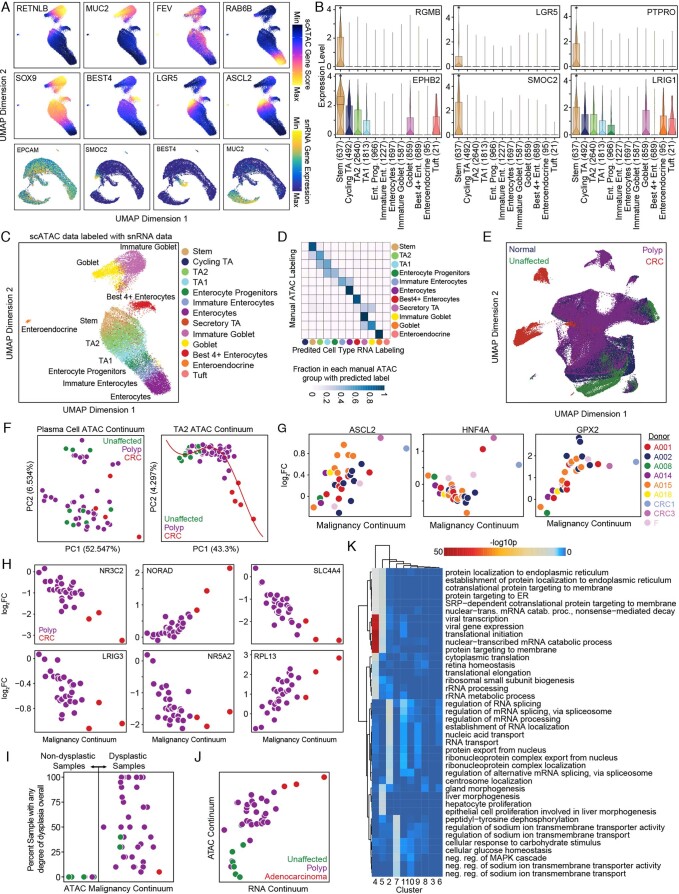
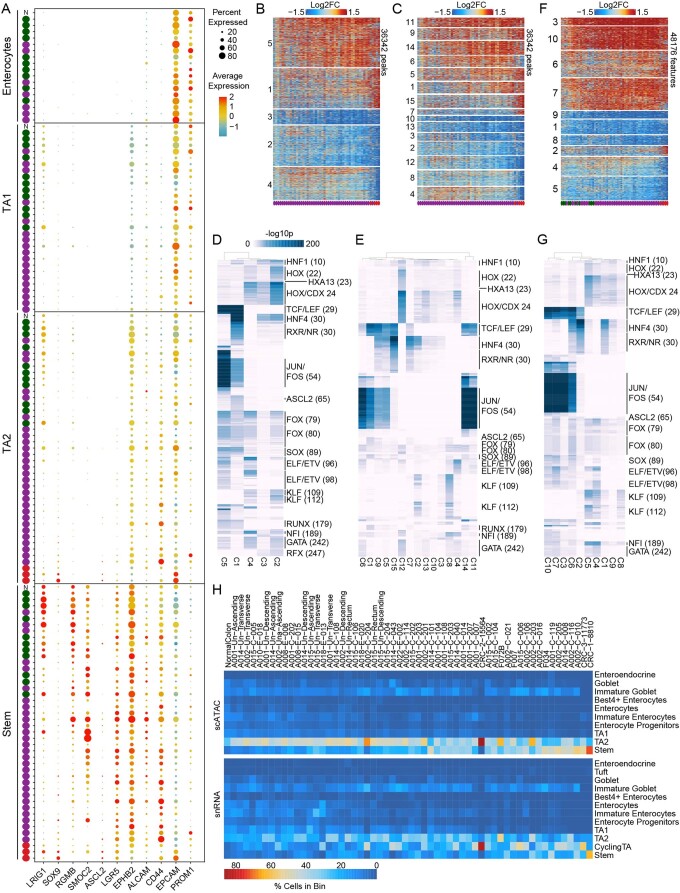
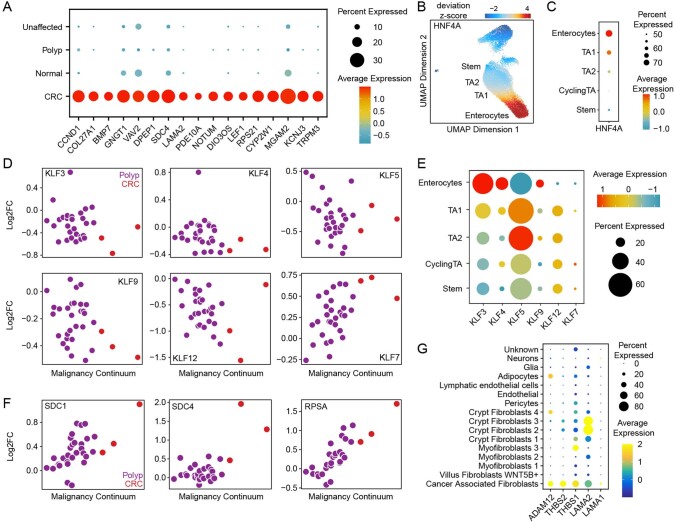
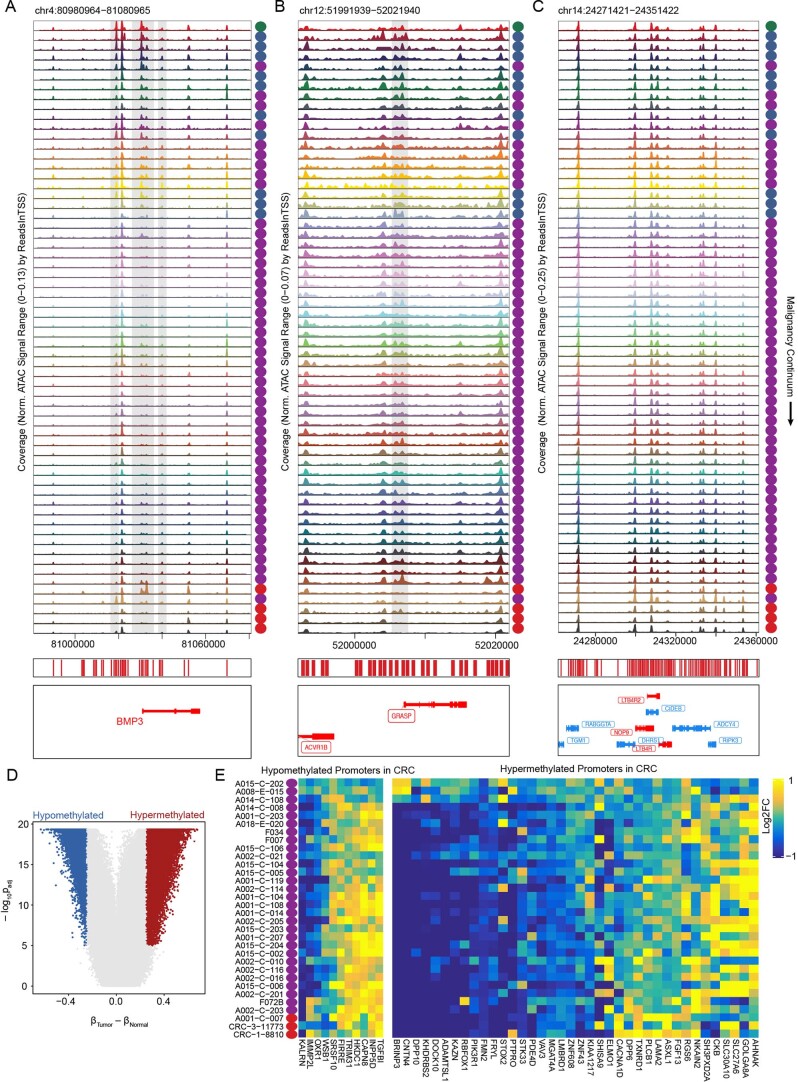
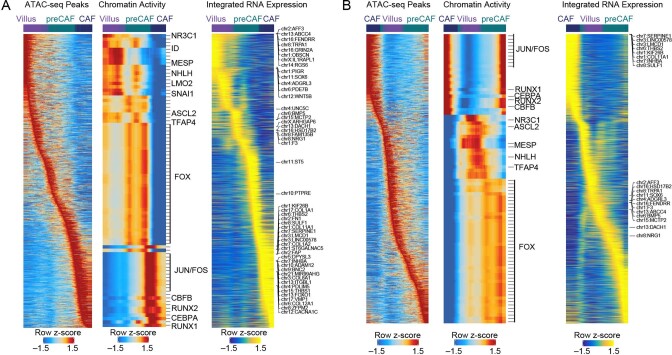
To chart cell composition and cell state changes that occur during the transformation of healthy colon to precancerous adenomas to colorectal cancer (CRC), we generated single-cell chromatin accessibility profiles and single-cell transcriptomes from 1,000 to 10,000 cells per sample for 48 polyps, 27 normal tissues and 6 CRCs collected from patients with or without germline APC mutations. A large fraction of polyp and CRC cells exhibit a stem-like phenotype, and we define a continuum of epigenetic and transcriptional changes occurring in these stem-like cells as they progress from homeostasis to CRC. Advanced polyps contain increasing numbers of stem-like cells, regulatory T cells and a subtype of pre-cancer-associated fibroblasts. In the cancerous state, we observe T cell exhaustion, RUNX1-regulated cancer-associated fibroblasts and increasing accessibility associated with HNF4A motifs in epithelia. DNA methylation changes in sporadic CRC are strongly anti-correlated with accessibility changes along this continuum, further identifying regulatory markers for molecular staging of polyps.
© 2022. The Author(s).
Conflict of interest statement
W.J.G. is a consultant and equity holder for 10x Genomics, Guardant Health, Quantapore and Ultima Genomics, and cofounder of Protillion Biosciences, and is named on patents describing ATAC-seq. M.P.S. is a cofounder and scientific advisor for Personalis, Qbio, January.ai, Filtricine, Mirvie and Protos, and an advisor for Genapsys. A.K. is a consultant with Illumina, Inc. The remaining authors declare no competing interests.
Figures
Comment in
-
Mapping the malignant transformation from polyps to CRC.Nat Rev Gastroenterol Hepatol. 2022 Aug;19(8):488. doi: 10.1038/s41575-022-00660-4. Nat Rev Gastroenterol Hepatol. 2022. PMID: 35778614 No abstract available.
References
-
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature578, 82–93 (2020).
-
- Fodde R, Smits R, Clevers H. APC, signal transduction and genetic instability in colorectal cancer. Nat. Rev. Cancer. 2001;1:55–67. - PubMed
-
- Aoki K, Taketo MM. Adenomatous polyposis coli (APC): a multi-functional tumor suppressor gene. J. Cell Sci. 2007;120:3327–3335. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
