

Clonal heterogeneity and rates of specific chromosome gains are risk predictors in childhood high-hyperdiploid B-cell acute lymphoblastic leukemia
- PMID: 35726693
- PMCID: PMC9394234
- DOI: 10.1002/1878-0261.13276
Clonal heterogeneity and rates of specific chromosome gains are risk predictors in childhood high-hyperdiploid B-cell acute lymphoblastic leukemia
Abstract
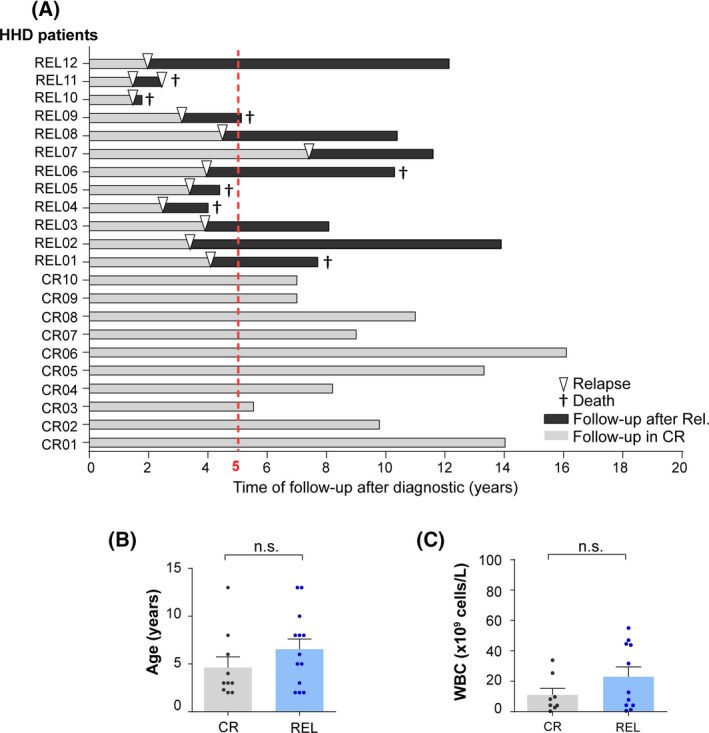
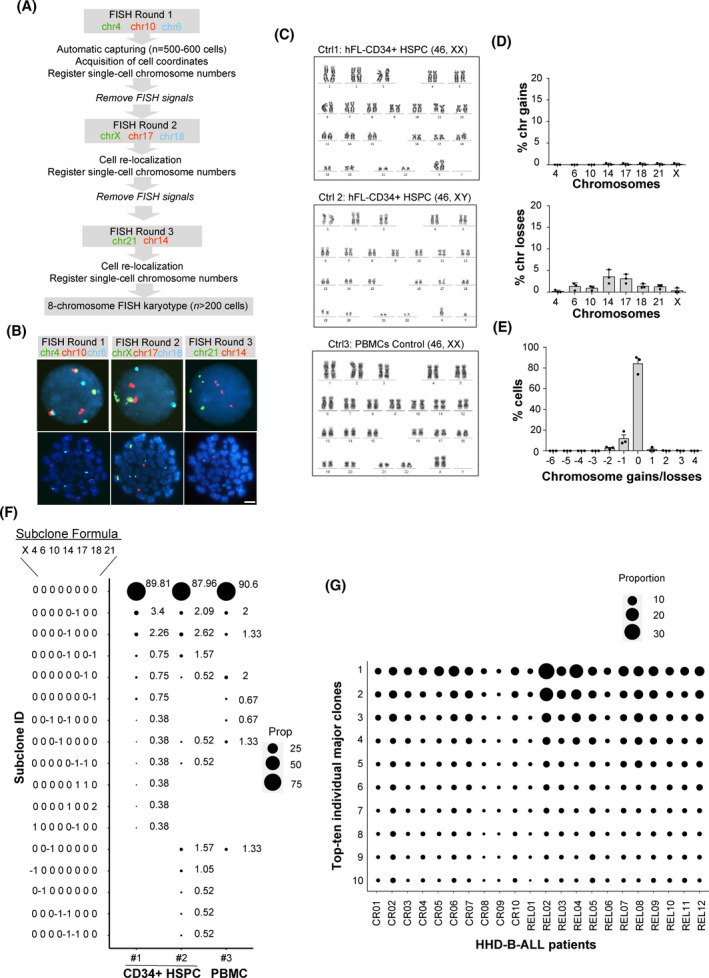
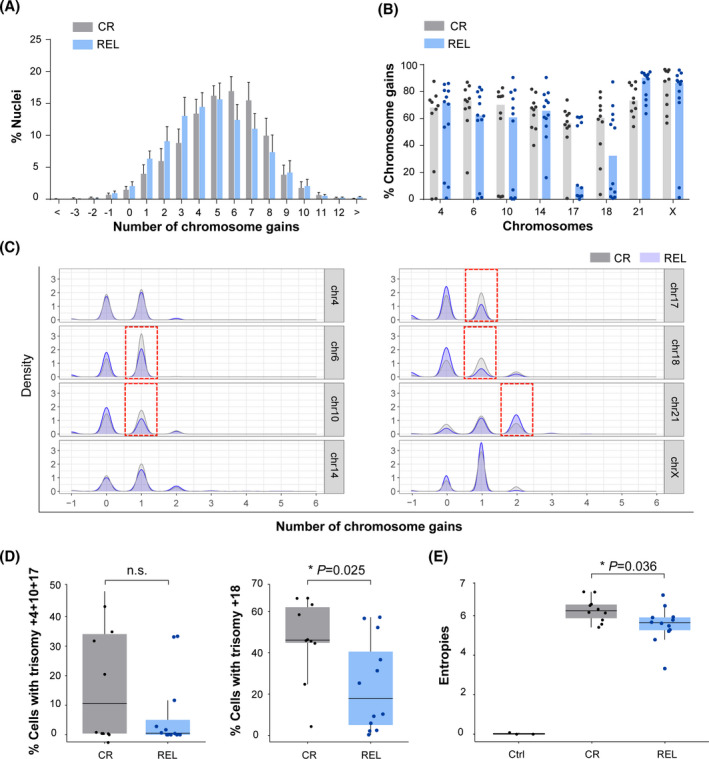
B-cell acute lymphoblastic leukemia (B-ALL) is the commonest childhood cancer. High hyperdiploidy (HHD) identifies the most frequent cytogenetic subgroup in childhood B-ALL. Although hyperdiploidy represents an important prognostic factor in childhood B-ALL, the specific chromosome gains with prognostic value in HHD-B-ALL remain controversial, and the current knowledge about the hierarchy of chromosome gains, clonal heterogeneity and chromosomal instability in HHD-B-ALL remains very limited. We applied automated sequential-iFISH coupled with single-cell computational modeling to identify the specific chromosomal gains of the eight typically gained chromosomes in a large cohort of 72 primary diagnostic (DX, n = 62) and matched relapse (REL, n = 10) samples from HHD-B-ALL patients with either favorable or unfavorable clinical outcome in order to characterize the clonal heterogeneity, specific chromosome gains and clonal evolution. Our data show a high degree of clonal heterogeneity and a hierarchical order of chromosome gains in DX samples of HHD-B-ALL. The rates of specific chromosome gains and clonal heterogeneity found in DX samples differ between HHD-B-ALL patients with favorable or unfavorable clinical outcome. In fact, our comprehensive analyses at DX using a computationally defined risk predictor revealed low levels of trisomies +18+10 and low levels of clonal heterogeneity as robust relapse risk factors in minimal residual disease (MRD)-negative childhood HHD-B-ALL patients: relapse-free survival beyond 5 years: 22.1% versus 87.9%, P < 0.0001 and 33.3% versus 80%, P < 0.0001, respectively. Moreover, longitudinal analysis of matched DX-REL HHD-B-ALL samples revealed distinct patterns of clonal evolution at relapse. Our study offers a reliable prognostic sub-stratification of pediatric MRD-negative HHD-B-ALL patients.
Keywords: chromosomal gains; clonal heterogeneity; computational modeling; high-hyperdiploid B-ALL; risk predictors; sequential iFISH.
© 2022 The Authors. Molecular Oncology published by John Wiley & Sons Ltd on behalf of Federation of European Biochemical Societies.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



References
-
- Schmiegelow K, Forestier E, Hellebostad M, Heyman M, Kristinsson J, Soderhall S, et al. Long‐term results of NOPHO ALL‐92 and ALL‐2000 studies of childhood acute lymphoblastic leukemia. Leukemia. 2010;24:345–54. - PubMed
