

Inhibition of myostatin and related signaling pathways for the treatment of muscle atrophy in motor neuron diseases
- PMID: 35727341
- PMCID: PMC9213329
- DOI: 10.1007/s00018-022-04408-w
Inhibition of myostatin and related signaling pathways for the treatment of muscle atrophy in motor neuron diseases
Abstract
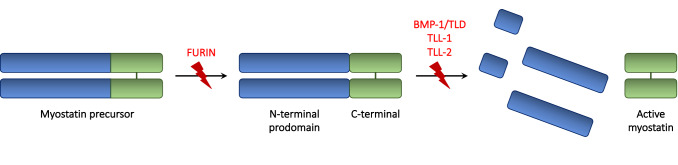
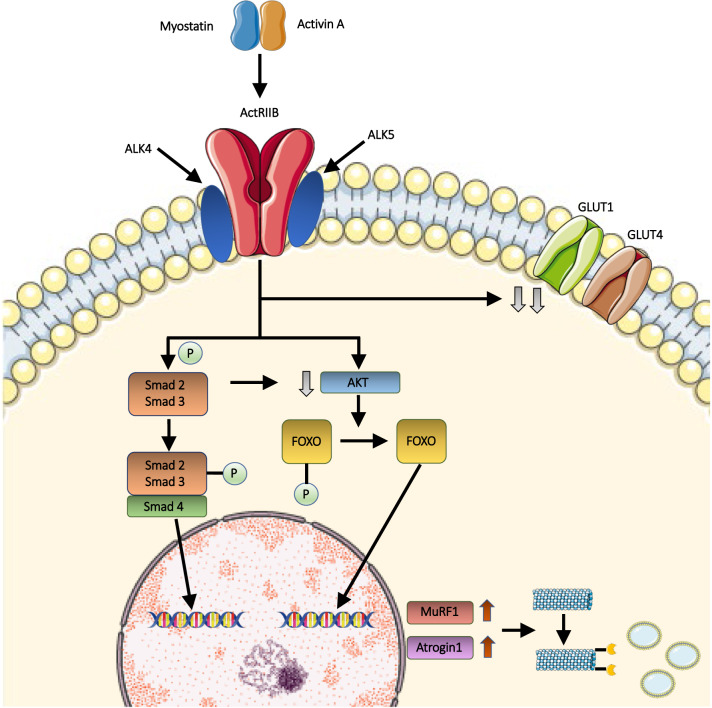
Myostatin is a negative regulator of skeletal muscle growth secreted by skeletal myocytes. In the past years, myostatin inhibition sparked interest among the scientific community for its potential to enhance muscle growth and to reduce, or even prevent, muscle atrophy. These characteristics make it a promising target for the treatment of muscle atrophy in motor neuron diseases, namely, amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA), which are rare neurological diseases, whereby the degeneration of motor neurons leads to progressive muscle loss and paralysis. These diseases carry a huge burden of morbidity and mortality but, despite this unfavorable scenario, several therapeutic advancements have been made in the past years. Indeed, a number of different curative therapies for SMA have been approved, leading to a revolution in the life expectancy and outcomes of SMA patients. Similarly, tofersen, an antisense oligonucleotide, is now undergoing clinical trial phase for use in ALS patients carrying the SOD1 mutation. However, these therapies are not able to completely halt or reverse progression of muscle damage. Recently, a trial evaluating apitegromab, a myostatin inhibitor, in SMA patients was started, following positive results from preclinical studies. In this context, myostatin inhibition could represent a useful strategy to tackle motor symptoms in these patients. The aim of this review is to describe the myostatin pathway and its role in motor neuron diseases, and to summarize and critically discuss preclinical and clinical studies of myostatin inhibitors in SMA and ALS. Then, we will highlight promises and pitfalls related to the use of myostatin inhibitors in the human setting, to aid the scientific community in the development of future clinical trials.
Keywords: Activin receptors, type II; Monoclonal antibodies; Motor neuron diseases; Muscle atrophy; Myostatin.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no existing conflict of interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
