

Targeting autophagy as a therapeutic strategy against pancreatic cancer
- PMID: 35727403
- PMCID: PMC9392712
- DOI: 10.1007/s00535-022-01889-1
Targeting autophagy as a therapeutic strategy against pancreatic cancer
Abstract
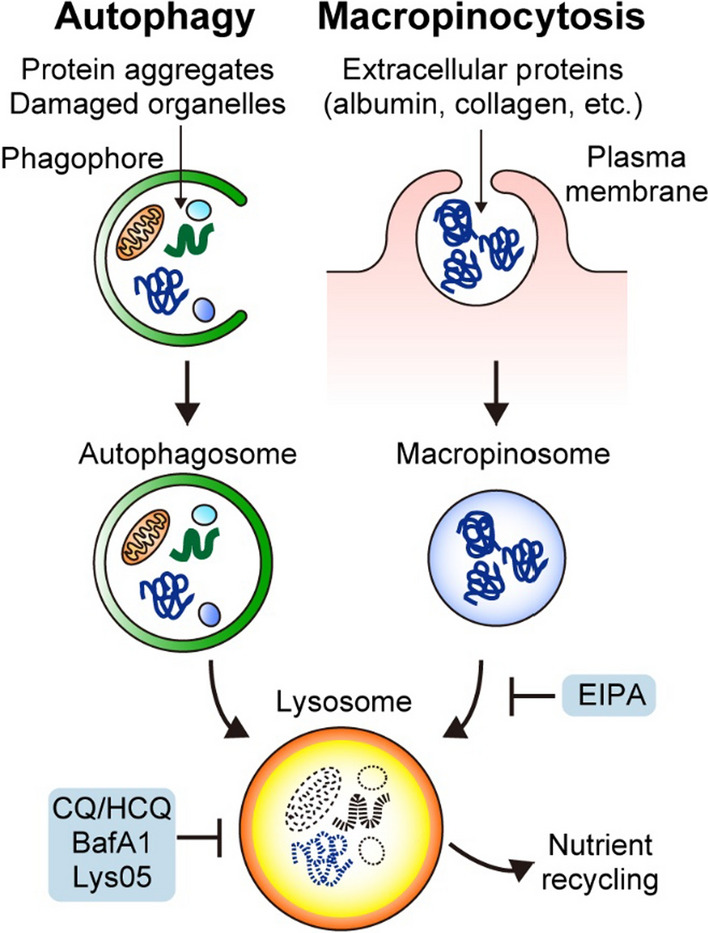
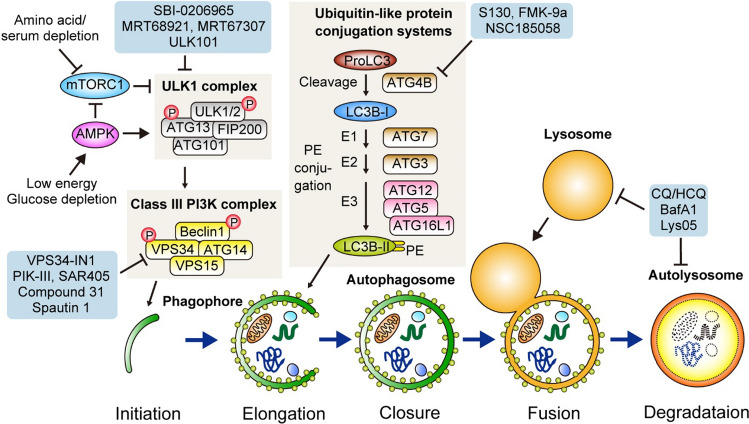
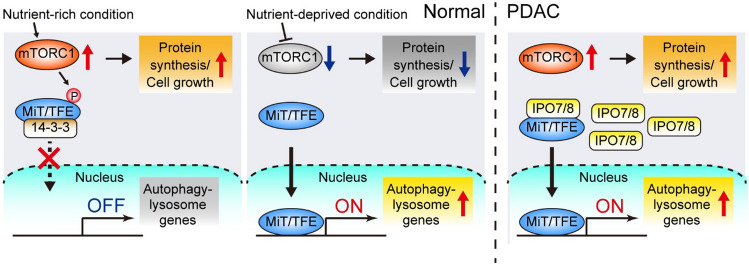
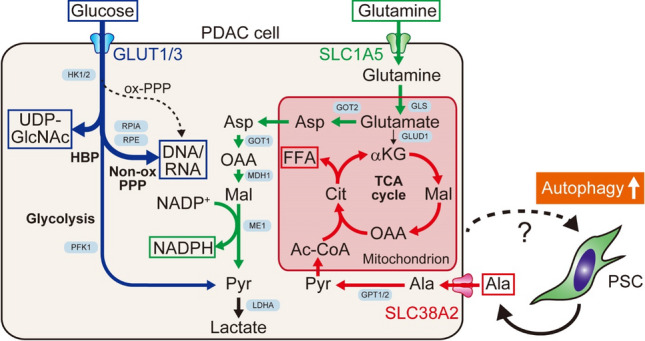
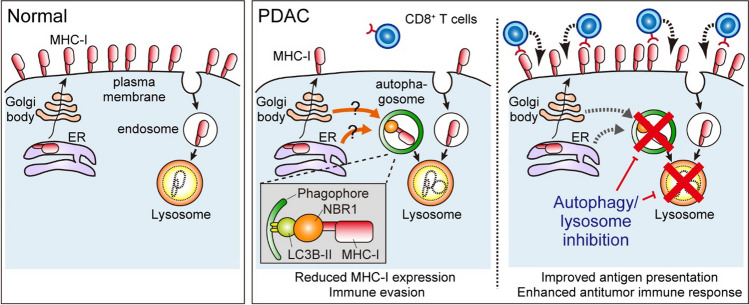
Macroautophagy (hereafter autophagy) is a catabolic process through which cytosolic components are captured in the autophagosome and degraded in the lysosome. Autophagy plays two major roles: nutrient recycling under starvation or stress conditions and maintenance of cellular homeostasis by removing the damaged organelles or protein aggregates. In established cancer cells, autophagy-mediated nutrient recycling promotes tumor progression, whereas in normal/premalignant cells, autophagy suppresses tumor initiation by eliminating the oncogenic/harmful molecules. Pancreatic ductal adenocarcinoma (PDAC) is a deadly disease that is refractory to most currently available treatment modalities, including immune checkpoint blockade and molecular-targeted therapy. One prominent feature of PDAC is its constitutively active and elevated autophagy-lysosome function, which enables PDAC to thrive in its nutrient-scarce tumor microenvironment. In addition to metabolic support, autophagy promotes PDAC progression in a metabolism-independent manner by conferring resistance to therapeutic treatment or facilitating immune evasion. Besides to cell-autonomous autophagy in cancer cells, host autophagy (autophagy in non-cancer cells) supports PDAC progression, further highlighting autophagy as a promising therapeutic target in PDAC. Based on a growing list of compelling preclinical evidence, there are numerous ongoing clinical trials targeting the autophagy-lysosome pathway in PDAC. Given the multifaceted and context-dependent roles of autophagy in both cancer cells and normal host cells, a deeper understanding of the mechanisms underlying the tumor-promoting roles of autophagy as well as of the consequences of autophagy inhibition is necessary for the development of autophagy inhibition-based therapies against PDAC.
Keywords: Anti-tumor immunity; Autophagy; Host autophagy; Lysosome; PDAC.
© 2022. The Author(s).
Conflict of interest statement
All authors declare that they have no conflicts of interest.
Figures
References
-
- Siegel RL, Miller KD, Fuchs HE, et al. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71:7–33. - PubMed
-
- Humphris JL, Patch AM, Nones K, et al. Hypermutation In pancreatic cancer. Gastroenterology. 2017;152(68–74):e2. - PubMed
-
- Luchini C, Bibeau F, Ligtenberg MJL, et al. ESMO recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with PD-1/PD-L1 expression and tumour mutational burden: a systematic review-based approach. Ann Oncol. 2019;30:1232–1243. doi: 10.1093/annonc/mdz116. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
