

The niche-specialist and age-related oral microbial ecosystem: crosstalk with host immune cells in homeostasis
- PMID: 35731208
- PMCID: PMC9455711
- DOI: 10.1099/mgen.0.000811
The niche-specialist and age-related oral microbial ecosystem: crosstalk with host immune cells in homeostasis
Abstract
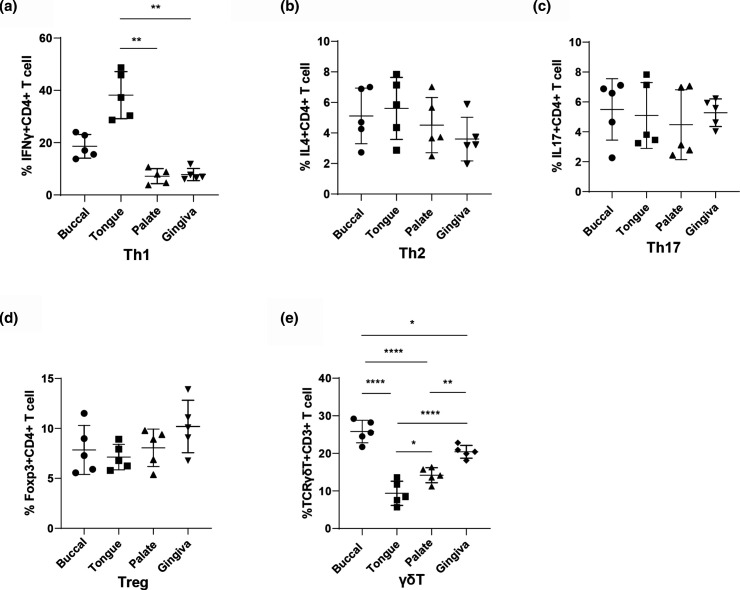
Although characterization of the baseline oral microbiota has been discussed, the current literature seems insufficient to draw a definitive conclusion on the interactions between the microbes themselves or with the host. This study focuses on the spatial and temporal characteristics of the oral microbial ecosystem in a mouse model and its crosstalk with host immune cells in homeostasis. The V3V4 regions of the 16S rRNA gene of 20 samples from four niches (tongue, buccal mucosa, keratinized gingiva and hard palate) and 10 samples from two life stages (adult and old) were analysed. Flow cytometry (FCM) was used to investigate the resident immune cells. The niche-specialist and age-related communities, characterized based on the microbiota structure, interspecies communications, microbial functions and interactions with immune cells, were addressed. The phylum Firmicutes was the major component in the oral community. The microbial community profiles at the genus level showed that the relative abundances of the genera Bacteroides, Lactobacillus and Porphyromonas were enriched in the gingiva. The abundance of the genera Streptococcus, Faecalibaculum and Veillonella was increased in palatal samples, while the abundance of Neisseria and Bradyrhizobium was enriched in buccal samples. The genera Corynebacterium, Stenotrophomonas, Streptococcus and Fusobacterium were proportionally enriched in old samples, while Prevotella and Lacobacillus were enriched in adult samples. Network analysis showed that the genus Lactobacillus performed as a central node in the buccal module, while in the gingiva module, the central nodes were Nesterenkonia and Hydrogenophilus. FCM showed that the proportion of Th1 cells in the tongue samples (38.18 % [27.03-49.34 %]) (mean [range]) was the highest. The proportion of γδT cells in the buccal mucosa (25.82 % [22.1-29.54 %]) and gingiva (20.42 % [18.31-22.53 %]) samples was higher (P<0.01) than those in the palate (14.18 % [11.69-16.67 %]) and tongue (9.38 % [5.38-13.37 %] samples. The proportion of Th2 (31.3 % [16.16-46.44 %]), Th17 (27.06 % [15.76-38.36 %]) and Treg (29.74 % [15.71-43.77 %]) cells in the old samples was higher than that in the adult samples (P<0.01). Further analysis of the interplays between the microbiomes and immune cells indicated that Th1 cells in the adult group, nd Th2, Th17 and Treg cells in the old group were the main immune factors strongly associated with the oral microbiota. For example, Th2, Th17 and Treg cells showed a significantly positive correlation with age-related microorganisms such as Sphingomonas, Streptococcus and Acinetobacter, while Th1 cells showed a negative correlation. Another positive correlation occurred between Th1 cells and several commensal microbiomes such as Lactobacillus, Jeotgalicoccus and Sporosarcina. Th2, Th17 and Treg cells showed the opposite trend. Together, our findings identify the niche-specialist and age-related characteristics of the oral microbial ecosystem and the potential associations between the microbiomes and the mucosal immune cells, providing critical insights into mucosal microbiology.
Keywords: age-related microbiota; mucosal microbiology; niche-specialist microbiome; oral homeostasis; oral microbial ecosystem.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures







References
-
- Bergmeier LA. Oral Mucosa in Health and Disease. 1st edn. Cham: Springer; 2018. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources