

KRASG12C-independent feedback activation of wild-type RAS constrains KRASG12C inhibitor efficacy
- PMID: 35732135
- PMCID: PMC9809542
- DOI: 10.1016/j.celrep.2022.110993
KRASG12C-independent feedback activation of wild-type RAS constrains KRASG12C inhibitor efficacy
Abstract
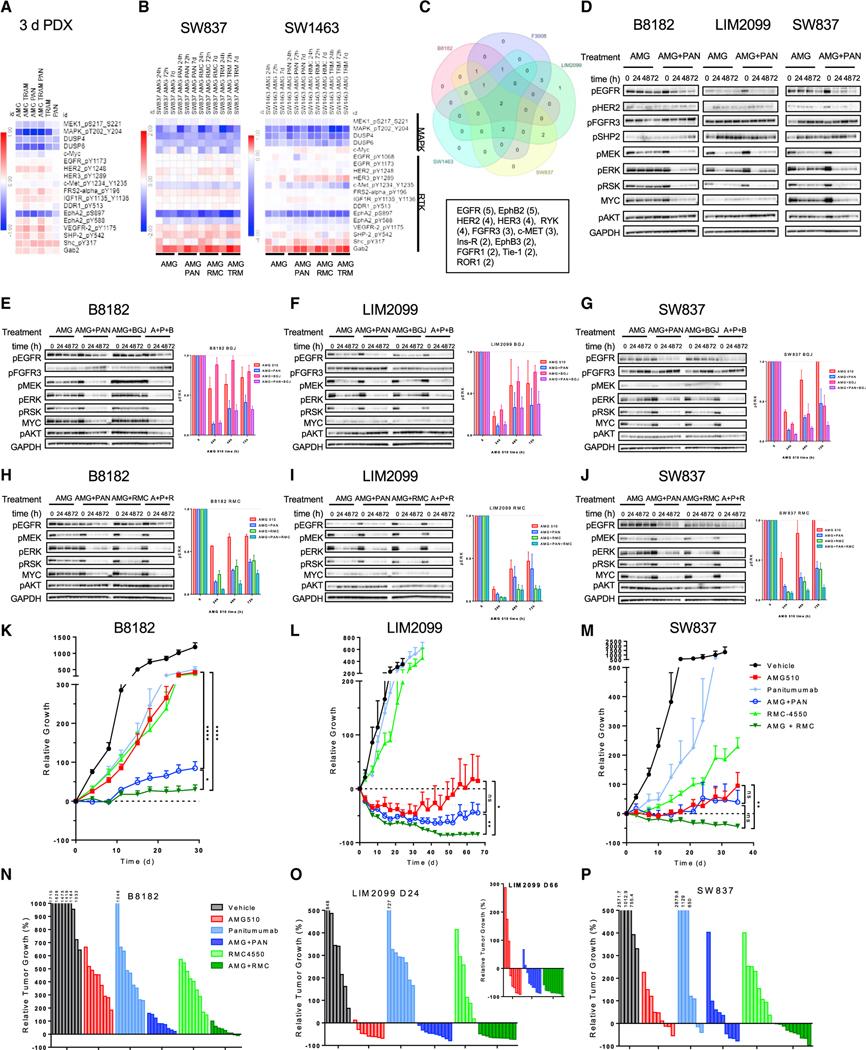
Although KRAS has long been considered undruggable, direct KRASG12C inhibitors have shown promising initial clinical efficacy. However, the majority of patients still fail to respond. Adaptive feedback reactivation of RAS-mitogen-activated protein kinase (MAPK) signaling has been proposed by our group and others as a key mediator of resistance, but the exact mechanism driving reactivation and the therapeutic implications are unclear. We find that upstream feedback activation of wild-type RAS, as opposed to a shift in KRASG12C to its active guanosine triphosphate (GTP)-bound state, is sufficient to drive RAS-MAPK reactivation in a KRASG12C-independent manner. Moreover, multiple receptor tyrosine kinases (RTKs) can drive feedback reactivation, potentially necessitating targeting of convergent signaling nodes for more universal efficacy. Even in colorectal cancer, where feedback is thought to be primarily epidermal growth factor receptor (EGFR)-mediated, alternative RTKs drive pathway reactivation and limit efficacy, but convergent upstream or downstream signal blockade can enhance activity. Overall, these data provide important mechanistic insight to guide therapeutic strategies targeting KRAS.
Keywords: CP: Cancer; KRAS; KRASG12C; adagrasib; adaptive resistance; sotorasib.
Copyright © 2022 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests R.B.C. has received consulting or speaking fees from Abbvie, Amgen, Array Biopharma/Pfizer, Asana Biosciences, Astex Pharmaceuticals, AstraZeneca, Avidity Biosciences, BMS, C4 Therapeutics, Chugai, Cogent Biosciences, Elicio, Erasca, Fog Pharma, Genentech, Guardant Health, Ipsen, Kinnate Biopharma, LOXO, Merrimack, Mirati Therapeutics, Natera, Navire, Nested Therapeutics, N-of-one/Qiagen, Novartis, nRichDx, Remix Therapeutics, Revolution Medicines, Roche, Roivant, Shionogi, Shire, Spectrum Pharmaceuticals, Symphogen, Syndax, Tango Therapeutics, Taiho, Theonys, Warp Drive Bio, and Zikani Therapeutics; holds equity in Alterome Therapeutics, Avidity Biosciences, C4 Therapeutics, Cogent Biosciences, Erasca, Kinnate Biopharma, Nested Therapeutics, nRichDx, Remix Therapeutics, Revolution Medicines, and Theonys; and has received research funding from Asana, AstraZeneca, Lilly, Novartis, and Sanofi. L.Z. and Y.Z. are employees and hold equity in Eli Lilly. SK. has received consulting or advisory fees from Genentech, EMD Serono, Merck, Holy Stone, Novartis, Lilly, Boehringer Ingelheim, Boston Biomedical, AstraZeneca/MedImmune, Bayer Health, Pierre Fabre, Redx Pharma, Ipsen, Daiichi Sankyo, Natera, HalioDx, Lutris, Jacobio, Pfizer, Repare Therapeutics, Inivata, GlaxoSmithKline, Jazz Pharmaceuticals, Iylon, Xilis, Abbvie, Amal Therapeutics, Gilead Sciences, Mirati Therapeutics, Flame Biosciences, Servier, Carina Biotechnology, Bicara Therapeutics, Endeavor BioMedicines, Numab Pharma, and Johnson & Johnson/Janssen.
Figures
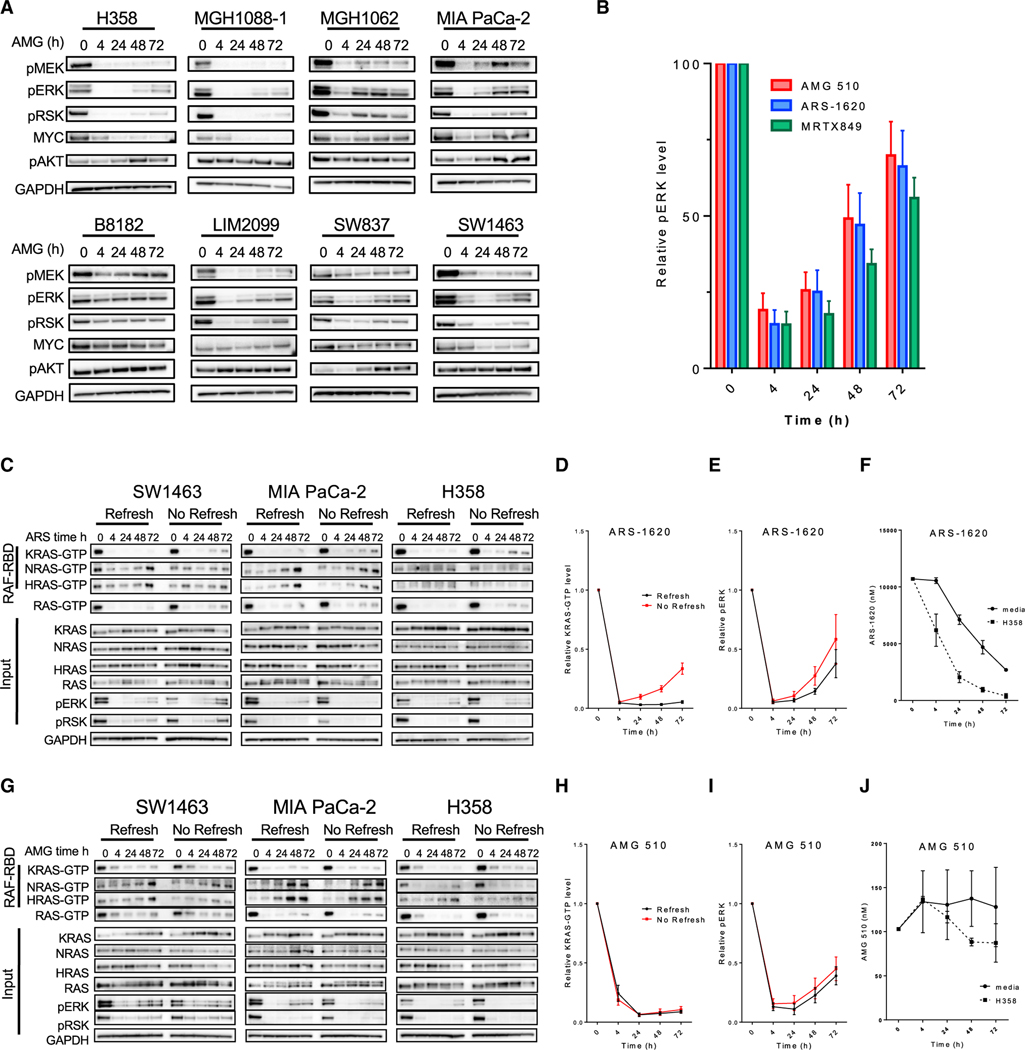
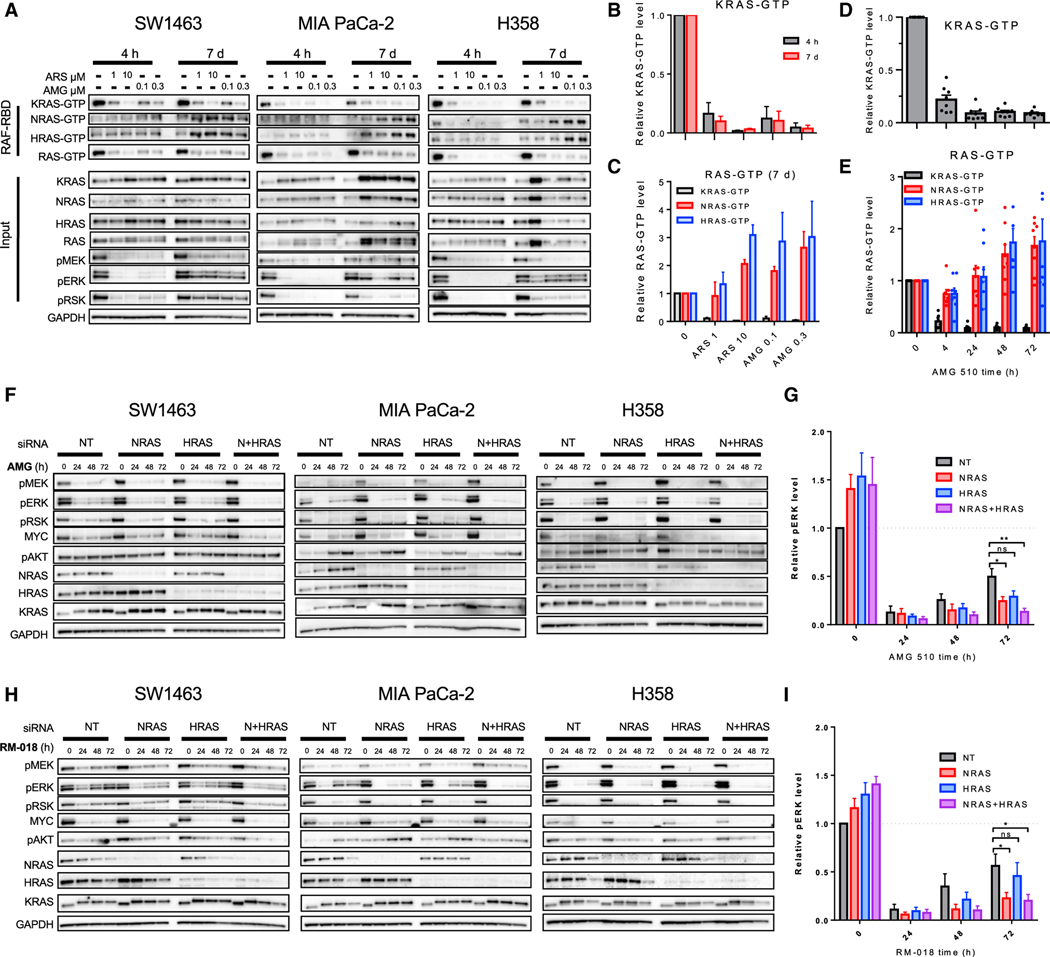
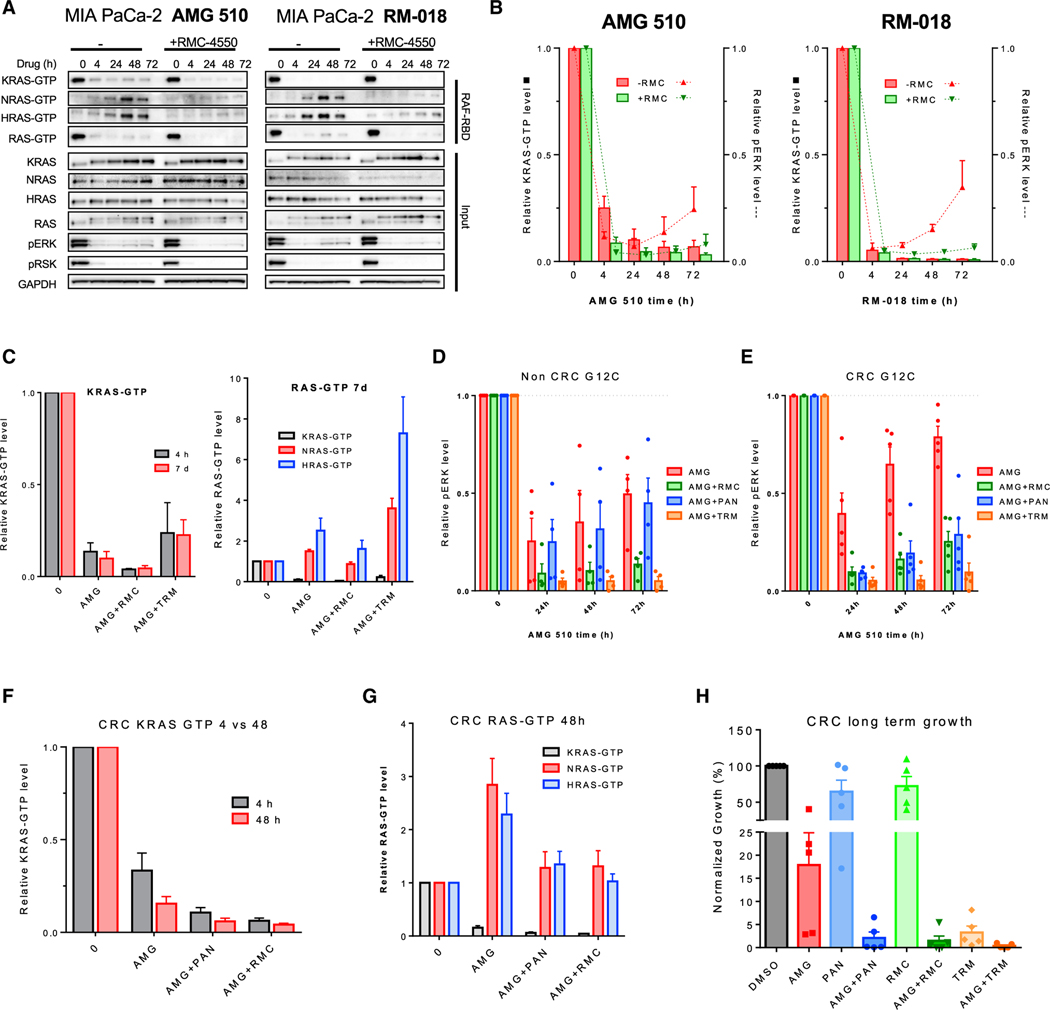
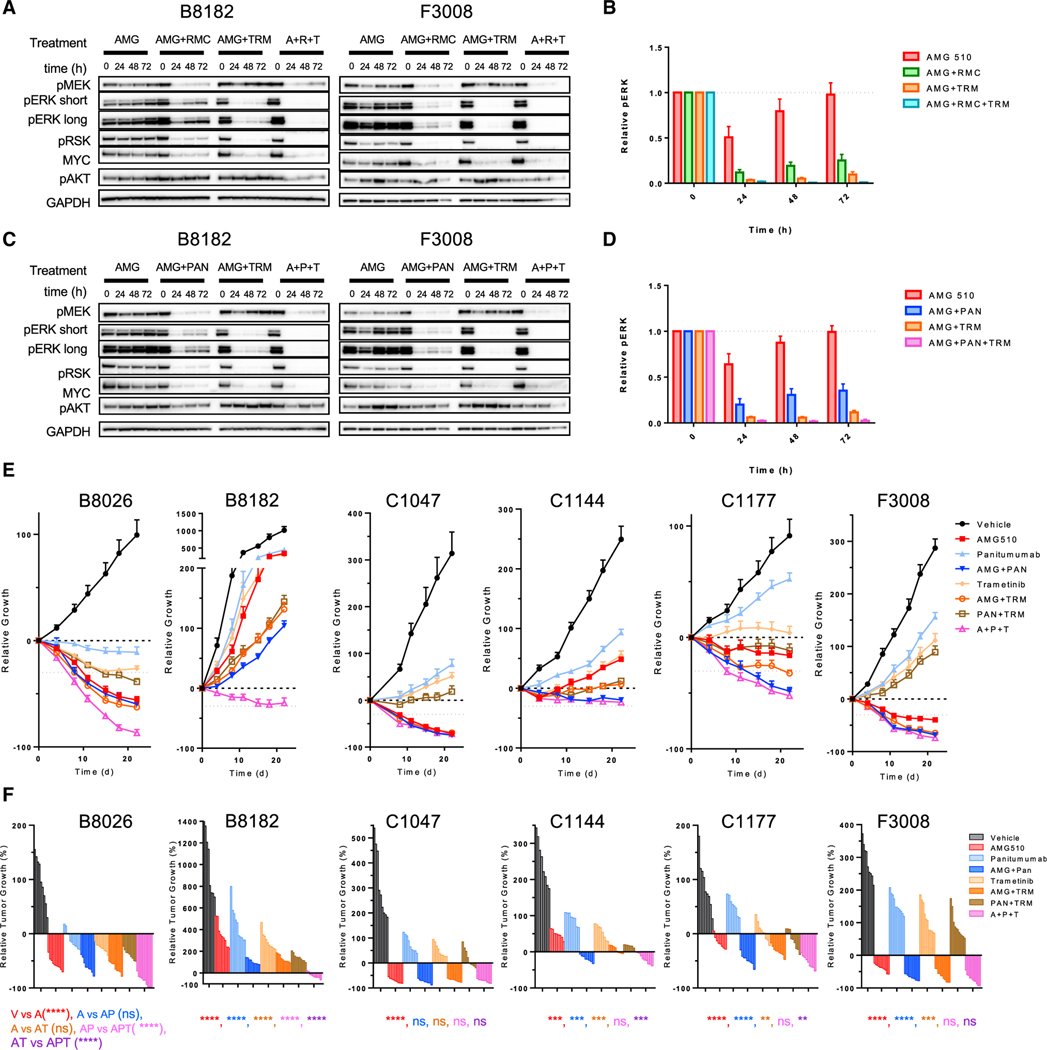
Comment in
-
KRASG12C mutation in metastatic colorectal cancer: a new target.Future Oncol. 2023 Aug;19(24):1641-1643. doi: 10.2217/fon-2023-0223. Epub 2023 Aug 21. Future Oncol. 2023. PMID: 37602398 No abstract available.
References
-
- Amodio V, Yaeger R, Arcella P, Cancelliere C, Lamba S, Lorenzato A, Arena S, Montone M, Mussolin B, Bian Y, et al. (2020). EGFR blockade reverts resistance to KRAS(G12C) inhibition in colorectal cancer. Cancer Discov. 10, 1129–1139. 10.1158/2159-8290.cd-20-0187. - DOI - PMC - PubMed
-
- Author Anonymous. (2020). Another KRAS Inhibitor Holds Its Own. Cancer Discov. 10, OF2. 10.1158/2159-8290.cd-nb2020-098. - DOI - PubMed
-
- Author Anonymous. (2021). FDA Approves First KRAS Inhibitor: Sotorasib. Cancer Discov. 11, OF4. 10.1158/2159-8290.cd-nb2021-0362. - DOI - PubMed
-
- Cheng DK, Oni TE, Thalappillil JS, Park Y, Ting HC, Alagesan B, Prasad NV, Addison K, Rivera KD, Pappin DJ, et al. (2021). Oncogenic KRAS engages an RSK1/NF1 pathway to inhibit wild-type RAS signaling in pancreatic cancer. Proc. Natl. Acad. Sci. U S A 118, e2016904118. 10.1073/pnas.2016904118. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
