

Optimal target saturation of ligand-blocking anti-GITR antibody IBI37G5 dictates FcγR-independent GITR agonism and antitumor activity
- PMID: 35732156
- PMCID: PMC9245059
- DOI: 10.1016/j.xcrm.2022.100660
Optimal target saturation of ligand-blocking anti-GITR antibody IBI37G5 dictates FcγR-independent GITR agonism and antitumor activity
Abstract
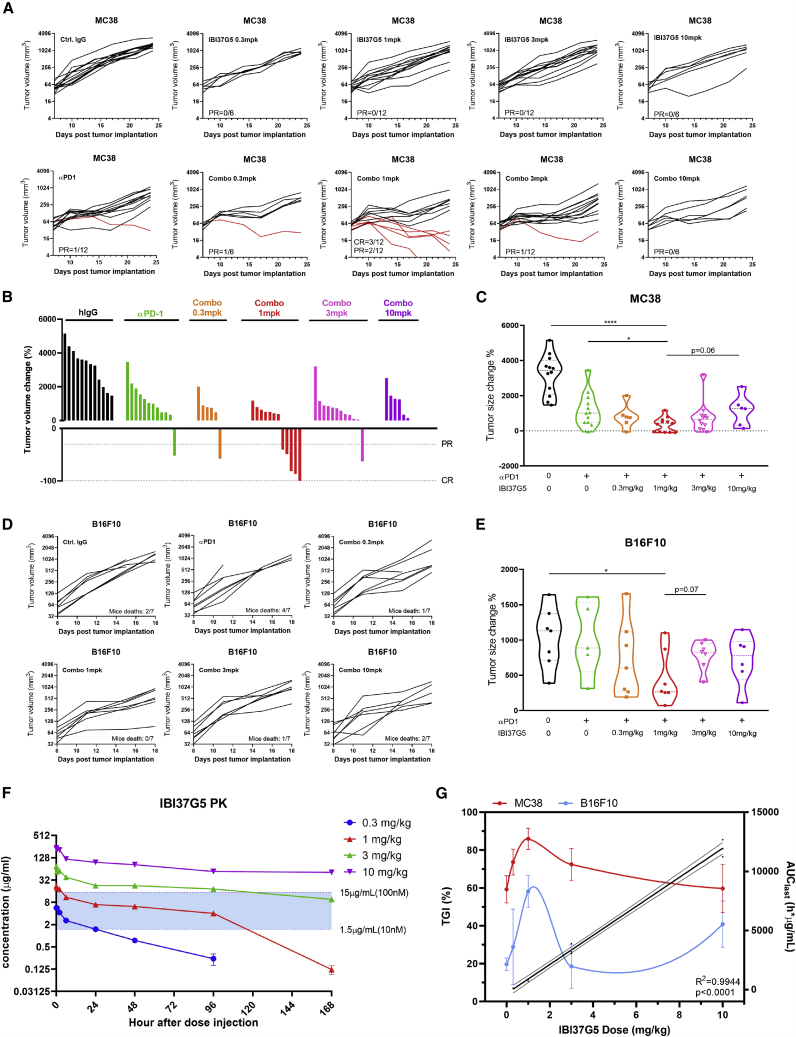
Glucocorticoid-induced tumor necrosis factor receptor (GITR) is a co-stimulatory receptor and an important target for cancer immunotherapy. We herein present a potent FcγR-independent GITR agonist IBI37G5 that can effectively activate effector T cells and synergize with anti-programmed death 1 (PD1) antibody to eradicate established tumors. IBI37G5 depends on both antibody bivalency and GITR homo-dimerization for efficient receptor cross-linking. Functional analyses reveal bell-shaped dose responses due to the unique 2:2 antibody-receptor stoichiometry required for GITR activation. Antibody self-competition is observed after concentration exceeded that of 100% receptor occupancy (RO), which leads to antibody monovalent binding and loss of activity. Retrospective pharmacokinetics/pharmacodynamics analysis demonstrates that the maximal efficacy is achieved at medium doses with drug exposure near saturating GITR occupancy during the dosing cycle. Finally, we propose an alternative dose-finding strategy that does not rely on the traditional maximal tolerated dose (MTD)-based paradigm but instead on utilizing the RO-function relations as biomarker to guide the clinical translation of GITR and similar co-stimulatory agonists.
Keywords: GITR; agonist antibody; cancer immunotherapy; costimulatory receptor; receptor occupancy.
Copyright © 2022 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests H.L., W.W., T.C., L.C., X.L., J.G., F.F., Y.Y., Z. Wu, S.Z., J.W., J.L., Z.K., M.W., Z.S., L.H., D.W., B.C., and K.H. are current employees and own stocks of Innovent Biologics (Suzhou) Co., Ltd. F.F. is co-inventor of GITR antibodies used in this study (patent no. WO2019201301A1).
Figures







Similar articles
-
Development of a fully human anti-GITR antibody with potent antitumor activity using H2L2 mice.FEBS Open Bio. 2022 Aug;12(8):1542-1557. doi: 10.1002/2211-5463.13451. Epub 2022 Jun 21. FEBS Open Bio. 2022. PMID: 35674216 Free PMC article.
-
HERA-GITRL activates T cells and promotes anti-tumor efficacy independent of FcγR-binding functionality.J Immunother Cancer. 2019 Jul 19;7(1):191. doi: 10.1186/s40425-019-0671-4. J Immunother Cancer. 2019. PMID: 31324216 Free PMC article.
-
An anti-PD-1-GITR-L bispecific agonist induces GITR clustering-mediated T cell activation for cancer immunotherapy.Nat Cancer. 2022 Mar;3(3):337-354. doi: 10.1038/s43018-022-00334-9. Epub 2022 Mar 7. Nat Cancer. 2022. PMID: 35256819 Free PMC article.
-
Role of the glucocorticoid-induced TNFR-related protein (GITR)-GITR ligand pathway in innate and adaptive immunity.Crit Rev Immunol. 2010;30(6):547-57. doi: 10.1615/critrevimmunol.v30.i6.40. Crit Rev Immunol. 2010. PMID: 21175417 Review.
-
T-cell intrinsic effects of GITR and 4-1BB during viral infection and cancer immunotherapy.Immunol Rev. 2011 Nov;244(1):197-217. doi: 10.1111/j.1600-065X.2011.01063.x. Immunol Rev. 2011. PMID: 22017440 Review.
Cited by
-
Engaging stimulatory immune checkpoint interactions in the tumour immune microenvironment of primary liver cancers - how to push the gas after having released the brake.Front Immunol. 2024 Feb 19;15:1357333. doi: 10.3389/fimmu.2024.1357333. eCollection 2024. Front Immunol. 2024. PMID: 38440738 Free PMC article. Review.
-
Novel hematopoietic progenitor kinase 1 inhibitor KHK-6 enhances T-cell activation.PLoS One. 2024 Jun 26;19(6):e0305261. doi: 10.1371/journal.pone.0305261. eCollection 2024. PLoS One. 2024. PMID: 38923962 Free PMC article.
References
-
- van Beek A.A., Zhou G., Doukas M., Boor P.P.C., Noordam L., Mancham S., Campos Carrascosa L., van der Heide-Mulder M., Polak W.G., Ijzermans J.N.M., et al. GITR ligation enhances functionality of tumor-infiltrating T cells in hepatocellular carcinoma. Int. J. Cancer. 2019;145:1111–1124. doi: 10.1002/ijc.32181. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
