

Cryo-EM structure of a type IV secretion system
- PMID: 35732732
- PMCID: PMC9259494
- DOI: 10.1038/s41586-022-04859-y
Cryo-EM structure of a type IV secretion system
Abstract
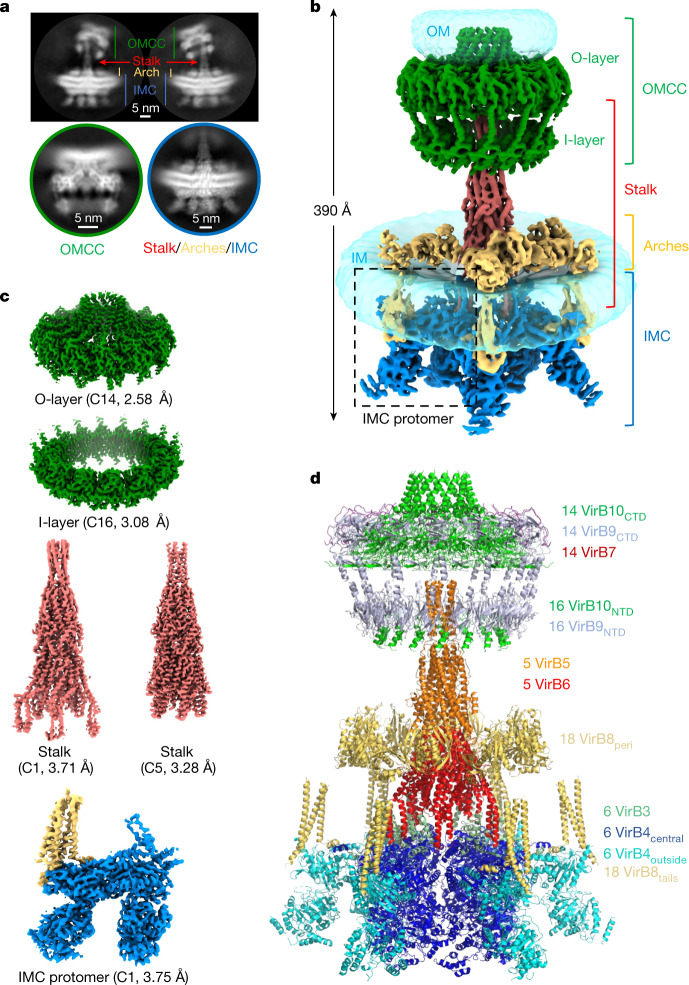
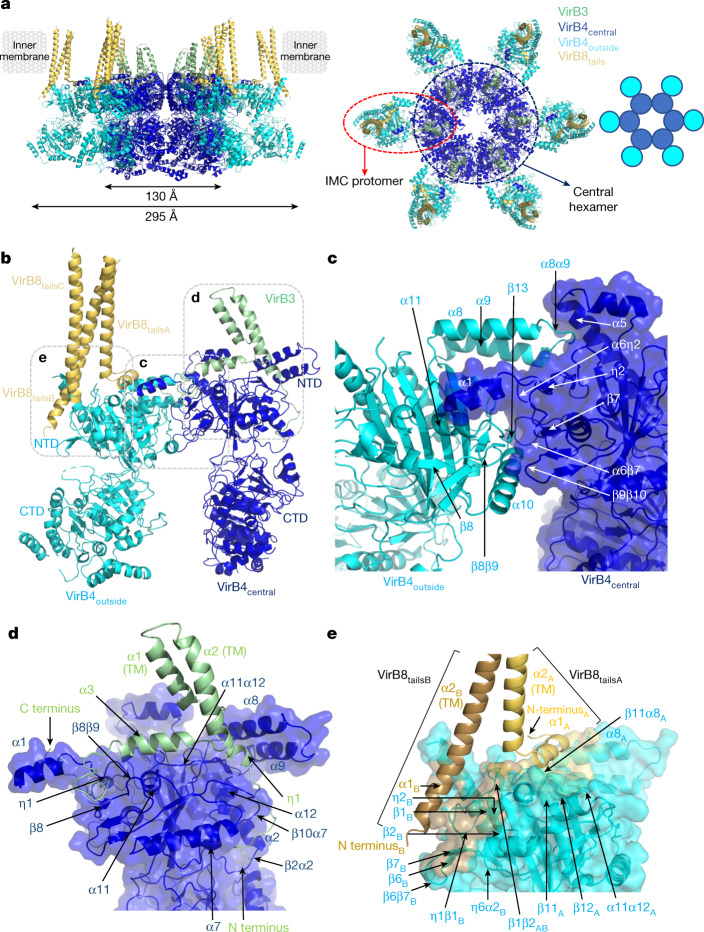
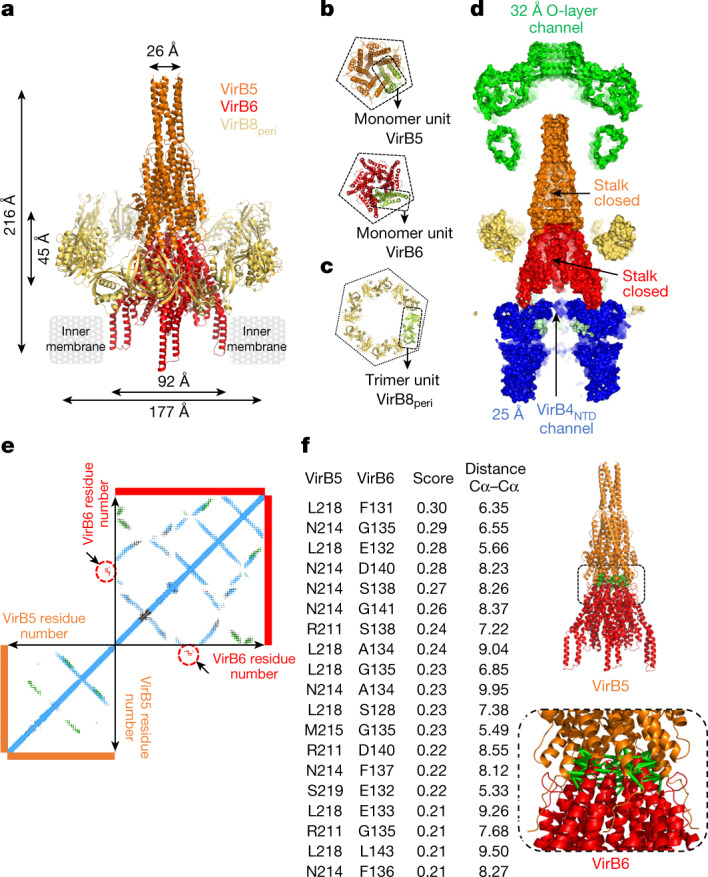
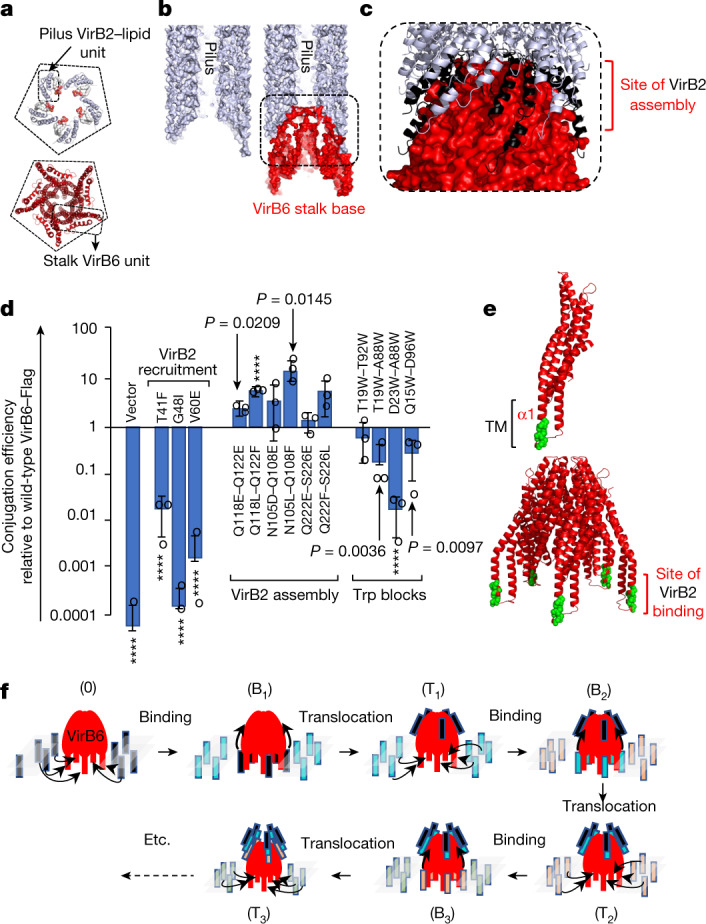
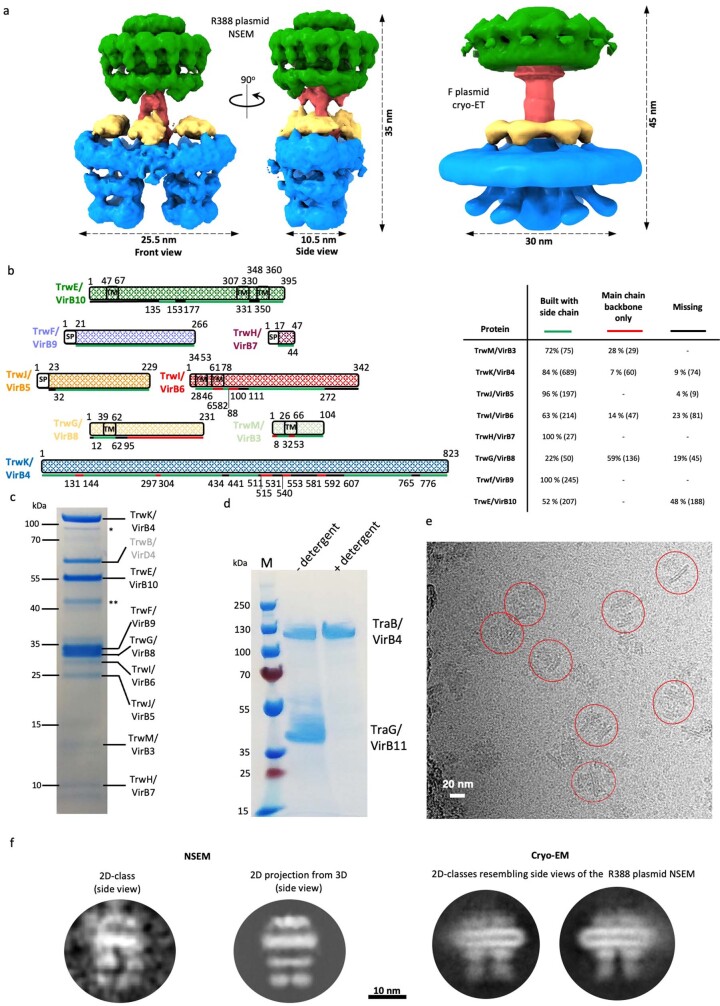
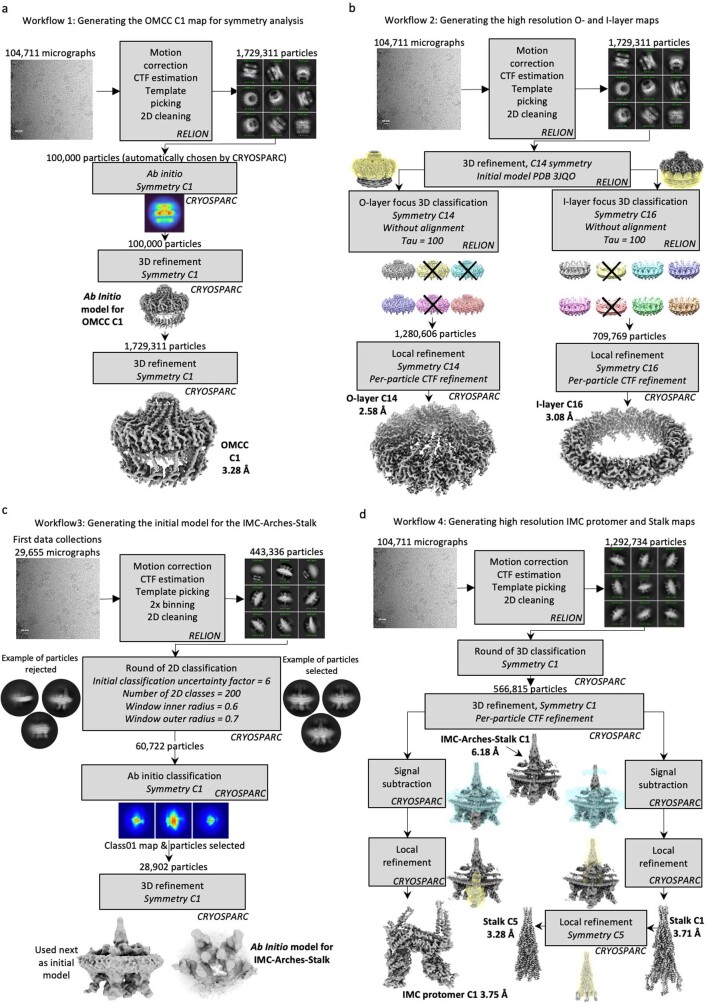
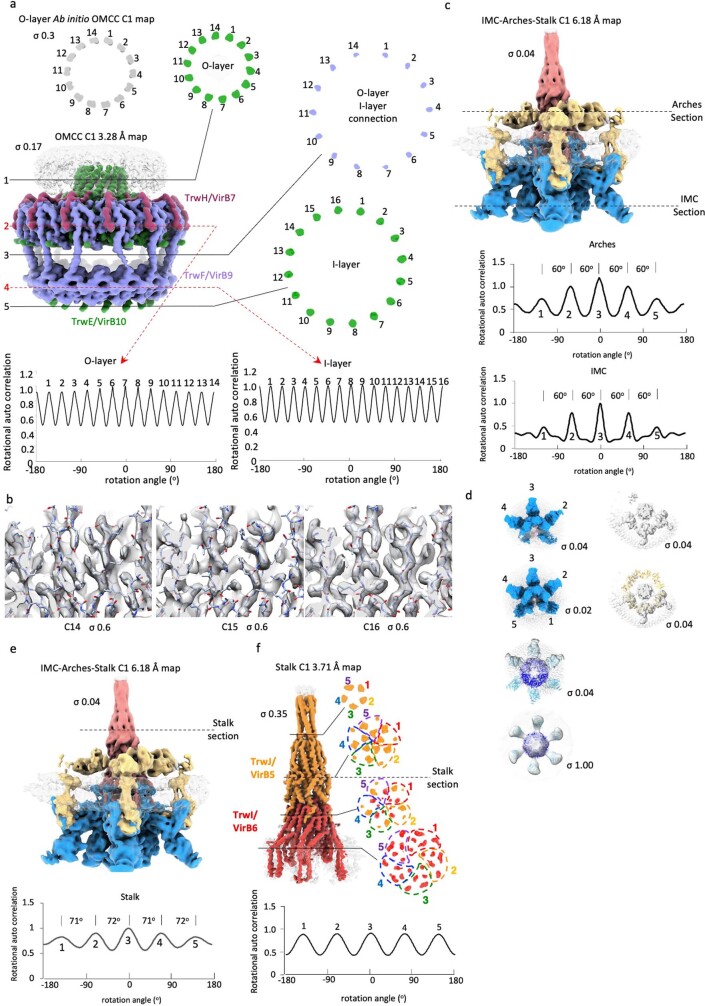
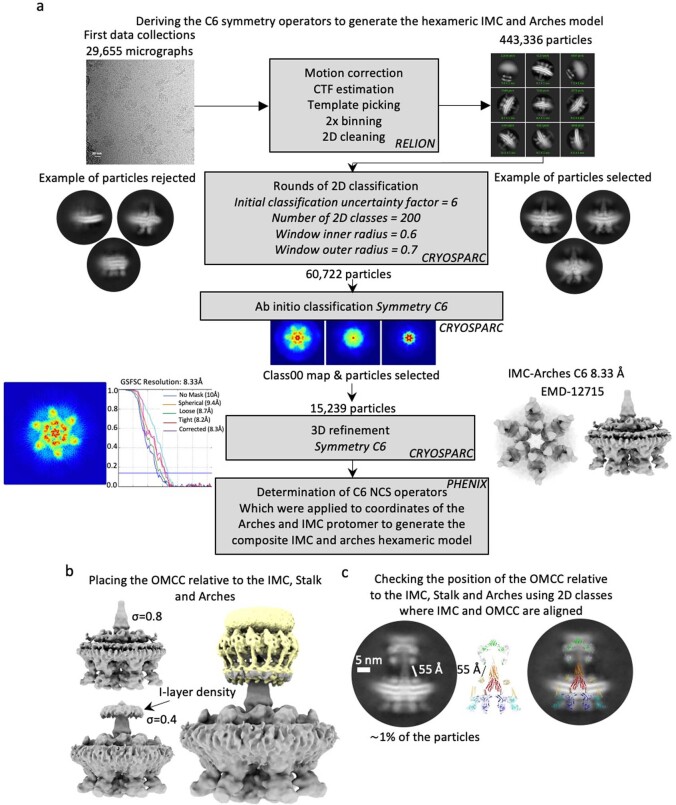
Bacterial conjugation is the fundamental process of unidirectional transfer of DNAs, often plasmid DNAs, from a donor cell to a recipient cell1. It is the primary means by which antibiotic resistance genes spread among bacterial populations2,3. In Gram-negative bacteria, conjugation is mediated by a large transport apparatus-the conjugative type IV secretion system (T4SS)-produced by the donor cell and embedded in both its outer and inner membranes. The T4SS also elaborates a long extracellular filament-the conjugative pilus-that is essential for DNA transfer4,5. Here we present a high-resolution cryo-electron microscopy (cryo-EM) structure of a 2.8 megadalton T4SS complex composed of 92 polypeptides representing 8 of the 10 essential T4SS components involved in pilus biogenesis. We added the two remaining components to the structural model using co-evolution analysis of protein interfaces, to enable the reconstitution of the entire system including the pilus. This structure describes the exceptionally large protein-protein interaction network required to assemble the many components that constitute a T4SS and provides insights on the unique mechanism by which they elaborate pili.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

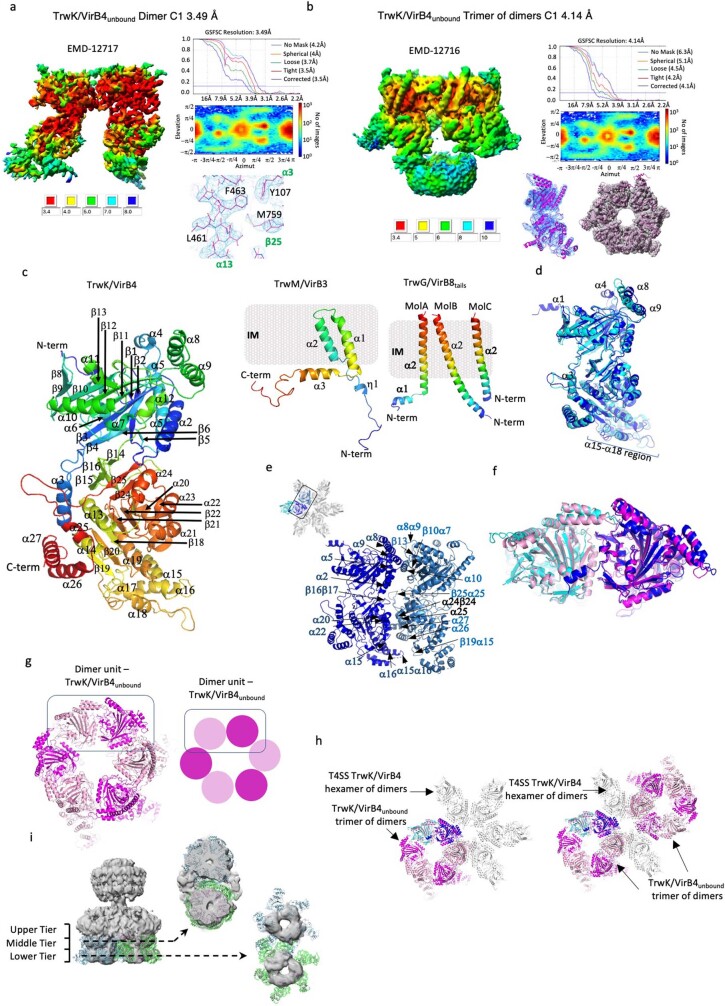
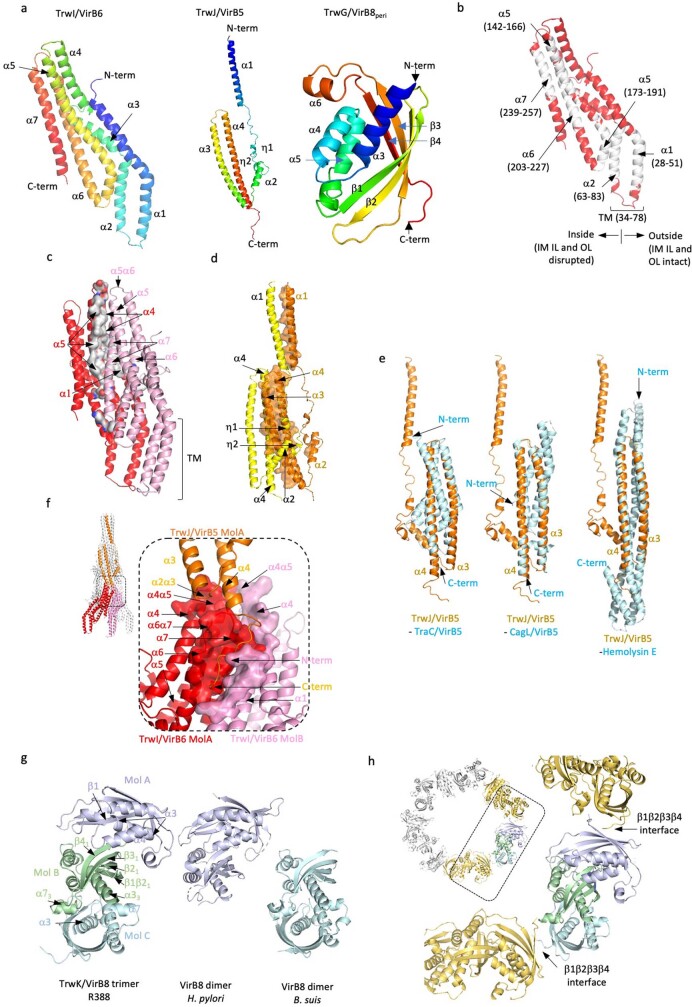
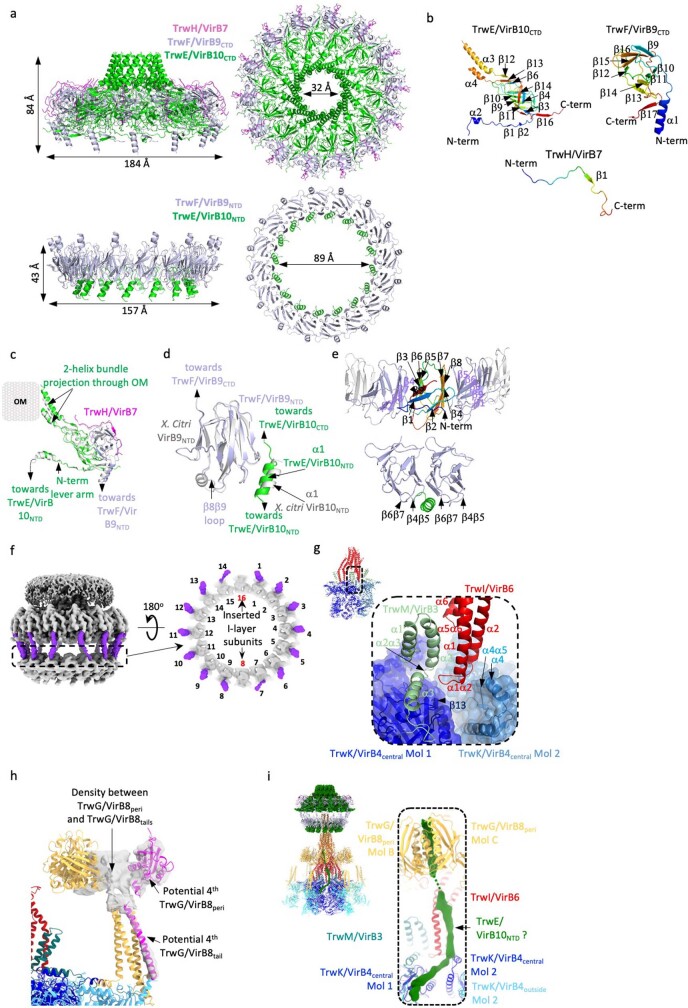
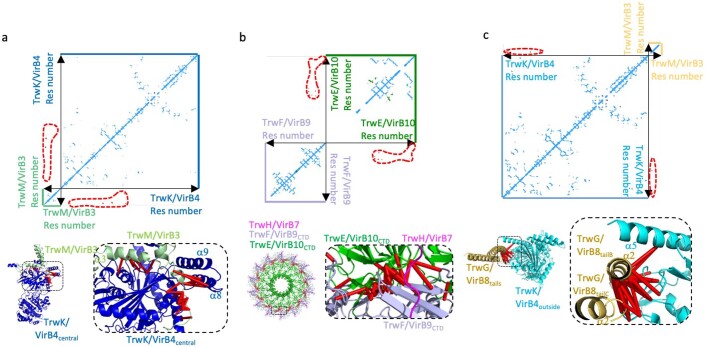
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
