

Biosynthetic potential of the global ocean microbiome
- PMID: 35732736
- PMCID: PMC9259500
- DOI: 10.1038/s41586-022-04862-3
Biosynthetic potential of the global ocean microbiome
Abstract
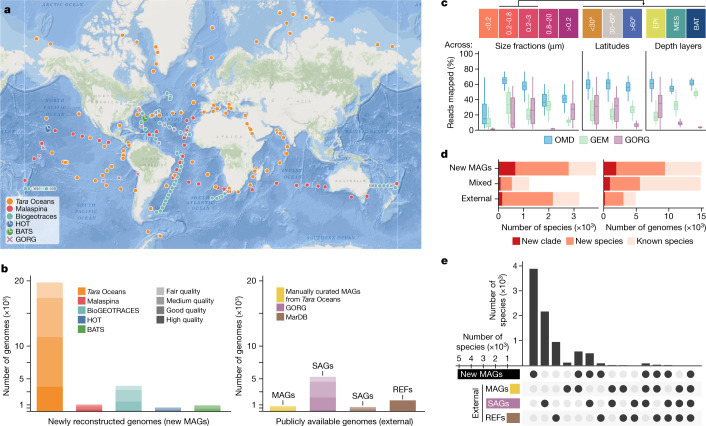
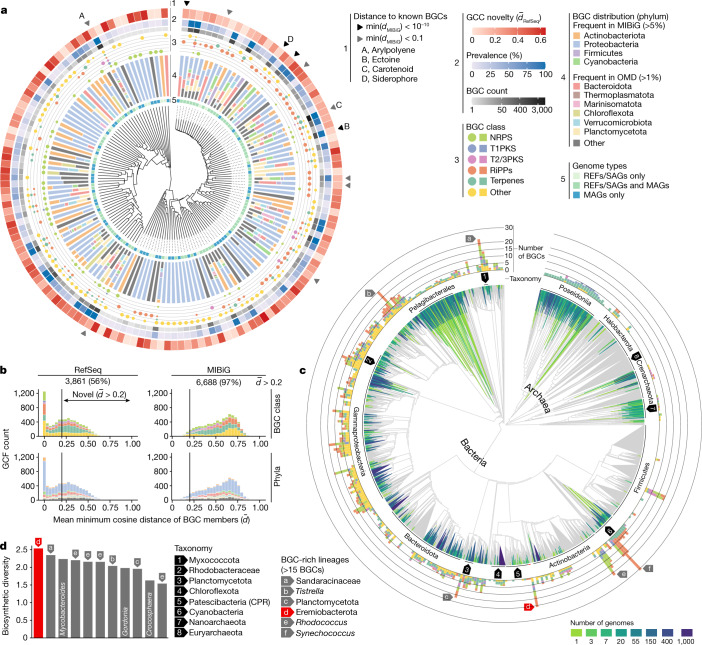
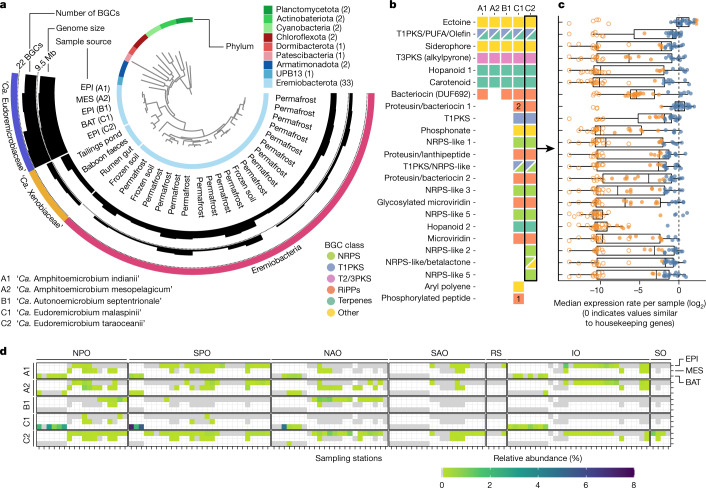
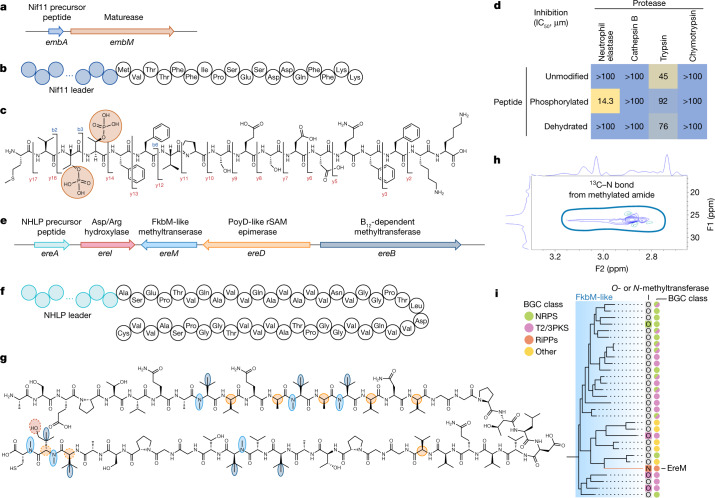
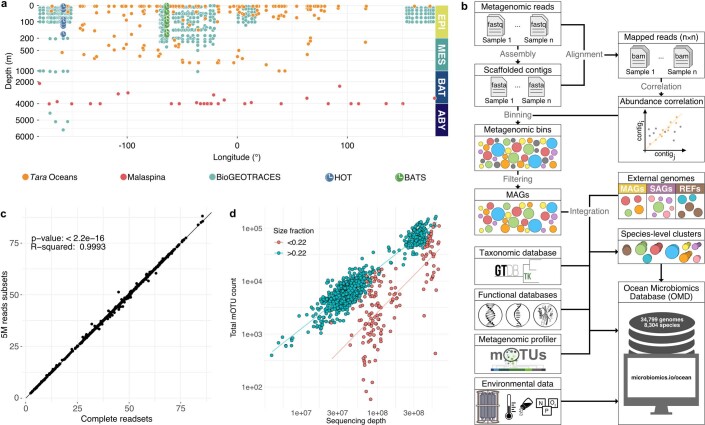
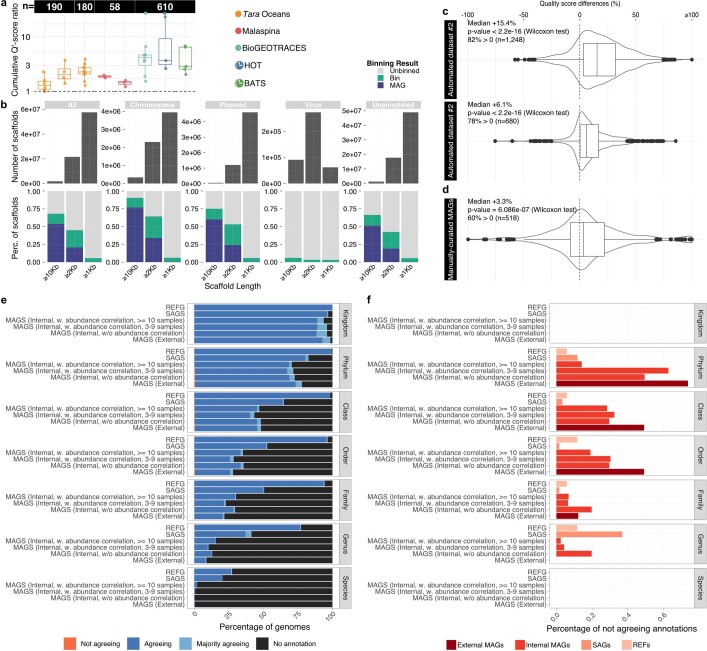
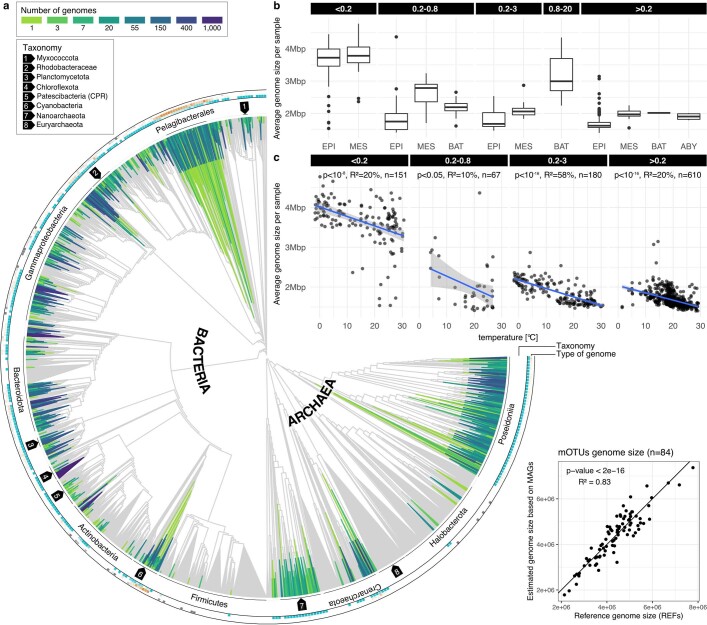
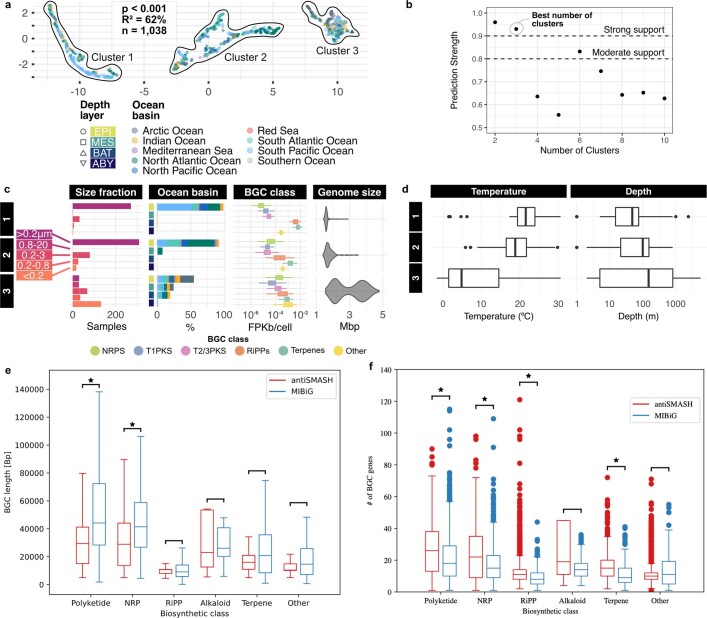
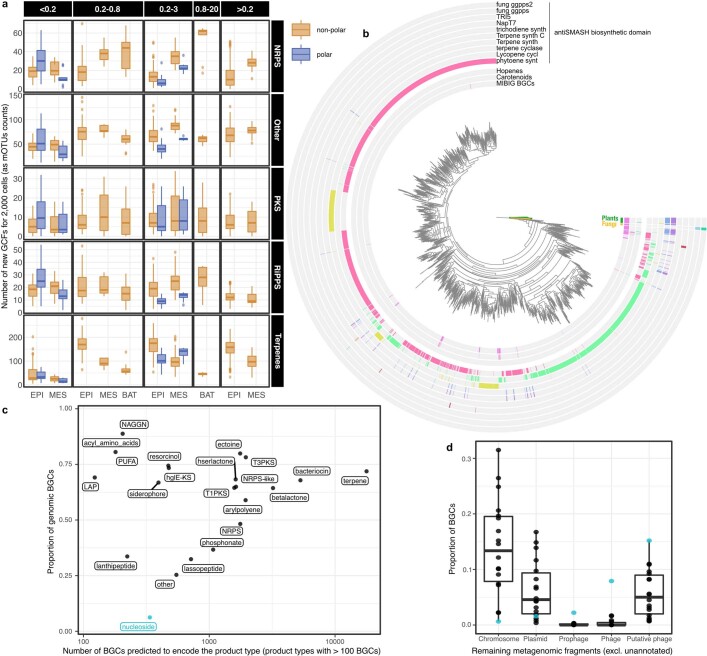
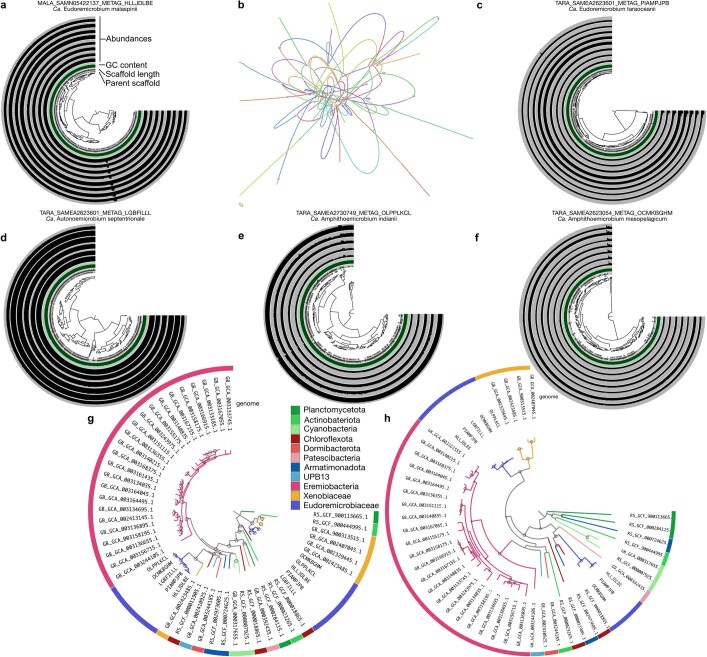
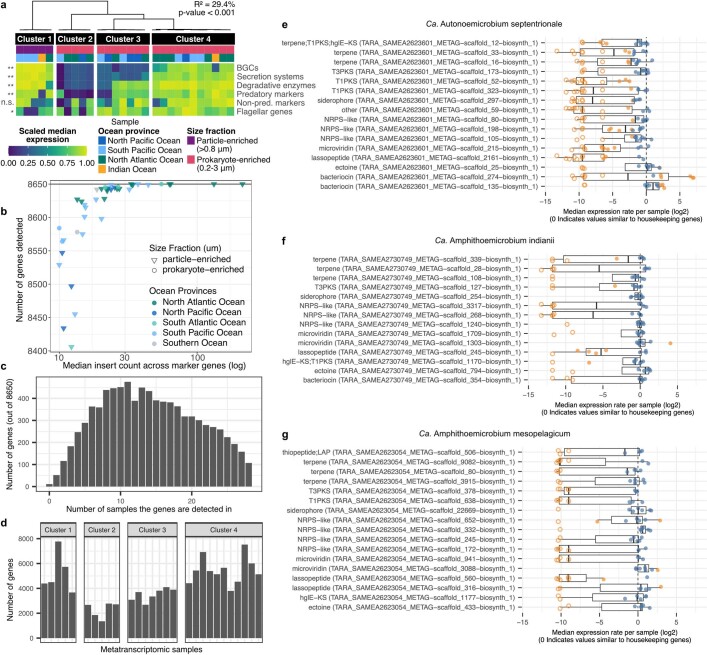
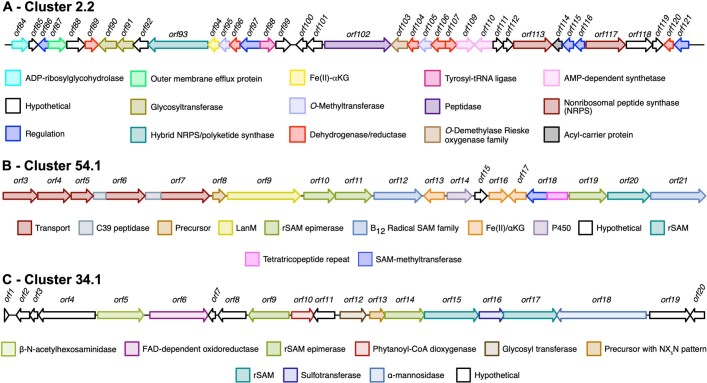
Natural microbial communities are phylogenetically and metabolically diverse. In addition to underexplored organismal groups1, this diversity encompasses a rich discovery potential for ecologically and biotechnologically relevant enzymes and biochemical compounds2,3. However, studying this diversity to identify genomic pathways for the synthesis of such compounds4 and assigning them to their respective hosts remains challenging. The biosynthetic potential of microorganisms in the open ocean remains largely uncharted owing to limitations in the analysis of genome-resolved data at the global scale. Here we investigated the diversity and novelty of biosynthetic gene clusters in the ocean by integrating around 10,000 microbial genomes from cultivated and single cells with more than 25,000 newly reconstructed draft genomes from more than 1,000 seawater samples. These efforts revealed approximately 40,000 putative mostly new biosynthetic gene clusters, several of which were found in previously unsuspected phylogenetic groups. Among these groups, we identified a lineage rich in biosynthetic gene clusters ('Candidatus Eudoremicrobiaceae') that belongs to an uncultivated bacterial phylum and includes some of the most biosynthetically diverse microorganisms in this environment. From these, we characterized the phospeptin and pythonamide pathways, revealing cases of unusual bioactive compound structure and enzymology, respectively. Together, this research demonstrates how microbiomics-driven strategies can enable the investigation of previously undescribed enzymes and natural products in underexplored microbial groups and environments.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
Charting the world's microbiomes.Nat Rev Genet. 2022 Sep;23(9):523. doi: 10.1038/s41576-022-00520-6. Nat Rev Genet. 2022. PMID: 35821096 No abstract available.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
