

Novel heterozygous variants of SLC12A6 in Japanese families with Charcot-Marie-Tooth disease
- PMID: 35733399
- PMCID: PMC9268887
- DOI: 10.1002/acn3.51603
Novel heterozygous variants of SLC12A6 in Japanese families with Charcot-Marie-Tooth disease
Abstract
Background: Recessive mutations in SLC12A6 have been linked to hereditary motor sensory neuropathy with agenesis of the corpus callosum. Patients with early-onset peripheral neuropathy associated with SLC12A6 heterozygous variants were reported in 2016. Only five families and three variants have been reported to date, and the spectrum is unclear. Here, we aim to describe the clinical and mutation spectra of SLC12A6-related Charcot-Marie-Tooth (CMT) disease in Japanese patients.
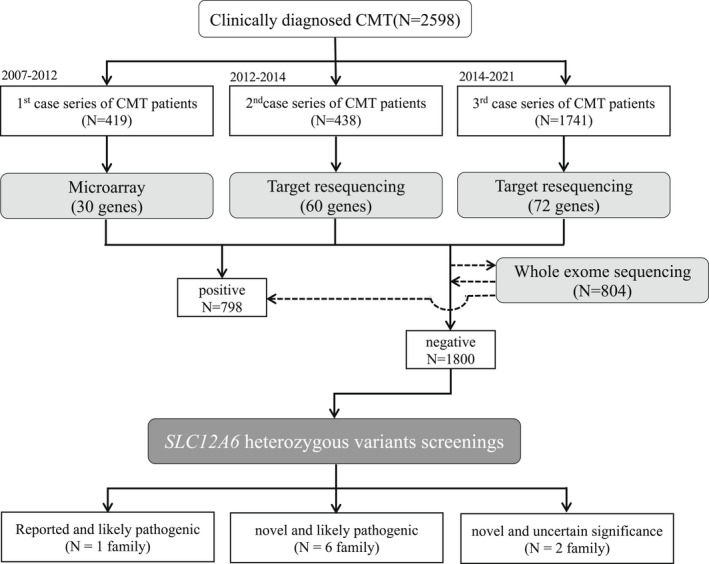
Methods: We extracted SLC12A6 variants from our DNA microarray and targeted resequencing data obtained from 2598 patients with clinically suspected CMT who were referred to our genetic laboratory by neurological or neuropediatric departments across Japan. And we summarized the clinical and genetic features of these patients.
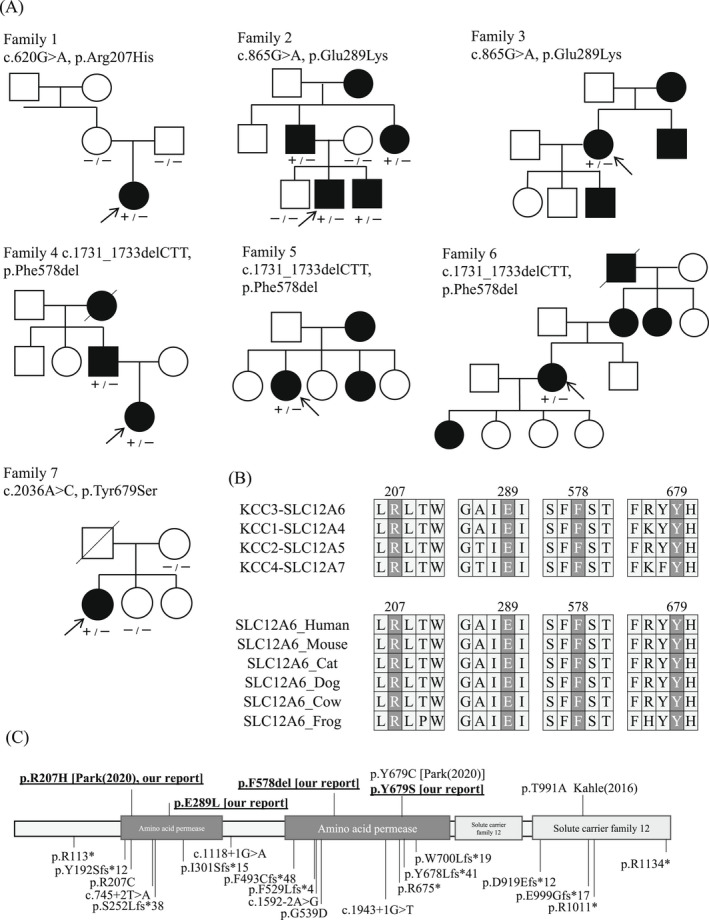
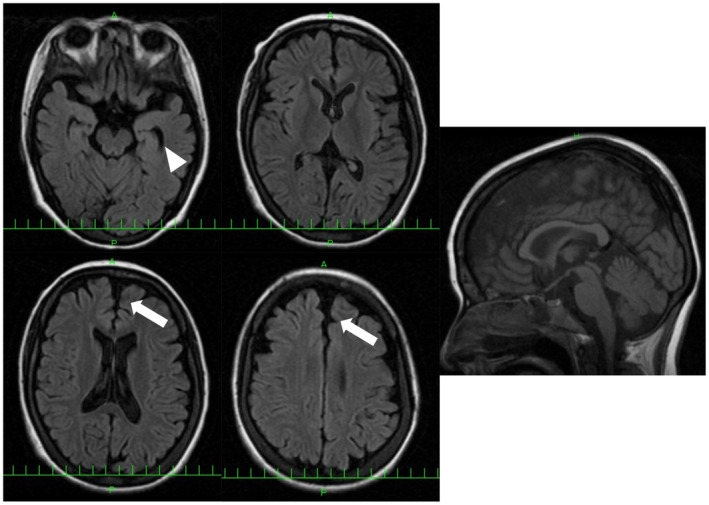
Results: In seven unrelated families, we identified one previously reported and three novel likely pathogenic SLC12A6 heterozygous variants, as well as two variants of uncertain significance. The mean age of onset for these patients was 17.5 ± 16.1 years. Regarding electrophysiology, the median motor nerve conduction velocity was 39.6 ± 9.5 m/sec. For the first time, we observed intellectual disability in three patients. One patient developed epilepsy, and her brain MRI revealed frontal and temporal lobe atrophy without changes in white matter and corpus callosum.
Conclusions: Screening for the SLC12A6 gene should be considered in patients with CMT, particularly those with central nervous system lesions, such as cognitive impairment and epilepsy, regardless of the CMT subtype.
© 2022 The Authors. Annals of Clinical and Translational Neurology published by Wiley Periodicals LLC on behalf of American Neurological Association.
Conflict of interest statement
All authors declare that there is no conflict of interest.
Figures
References
-
- Braathen GJ, Sand JC, Lobato A, Høyer H, Russell MB. Genetic epidemiology of Charcot‐Marie‐Tooth in the general population. Eur J Neurol. 2011;18:39‐48. - PubMed
-
- Stojkovic T. Hereditary neuropathies: an update. Rev Neurol (Paris). 2016;172(12):775‐778. - PubMed
-
- Zuchner S, Mersiyanova IV, Muglia M, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot‐Marie‐Tooth neuropathy type 2A. Nat Genet. 2004;36:449‐451. - PubMed
-
- Baxter RV, Ben Othmane K, Rochelle JM, et al. Ganglioside‐induced differentiation‐associated protein‐1 is mutant in Charcot‐Marie‐Tooth disease type 4A/8q21. Nat Genet. 2002;30:21‐22. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
