

Identification and Characterization of Major Bile Acid 7α-Dehydroxylating Bacteria in the Human Gut
- PMID: 35736002
- PMCID: PMC9426597
- DOI: 10.1128/msystems.00455-22
Identification and Characterization of Major Bile Acid 7α-Dehydroxylating Bacteria in the Human Gut
Abstract
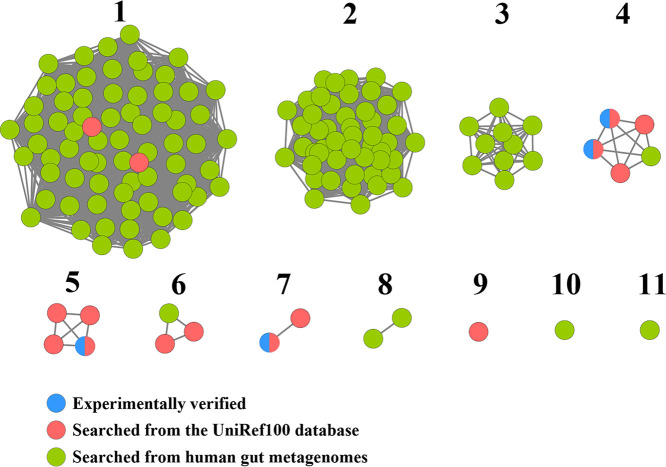
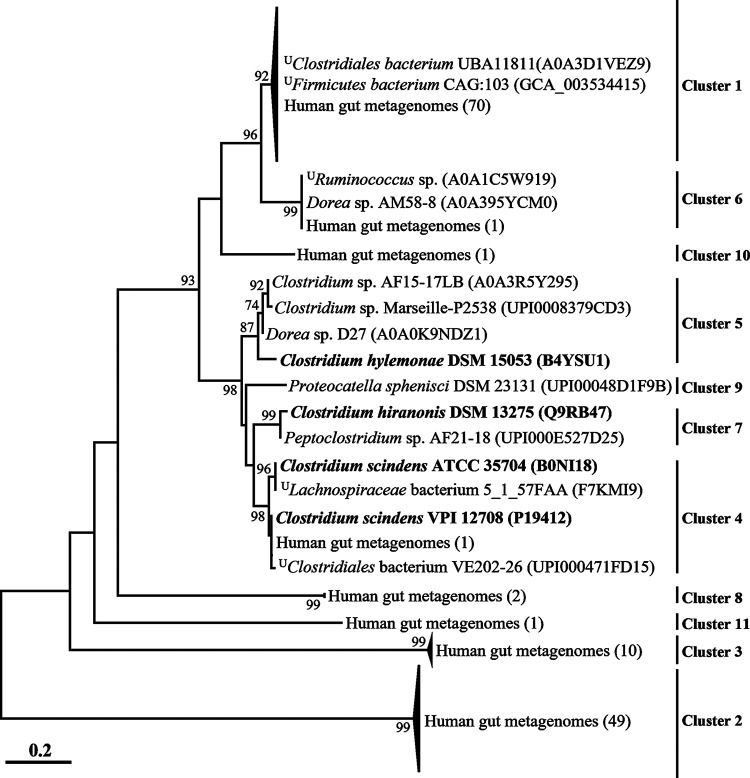
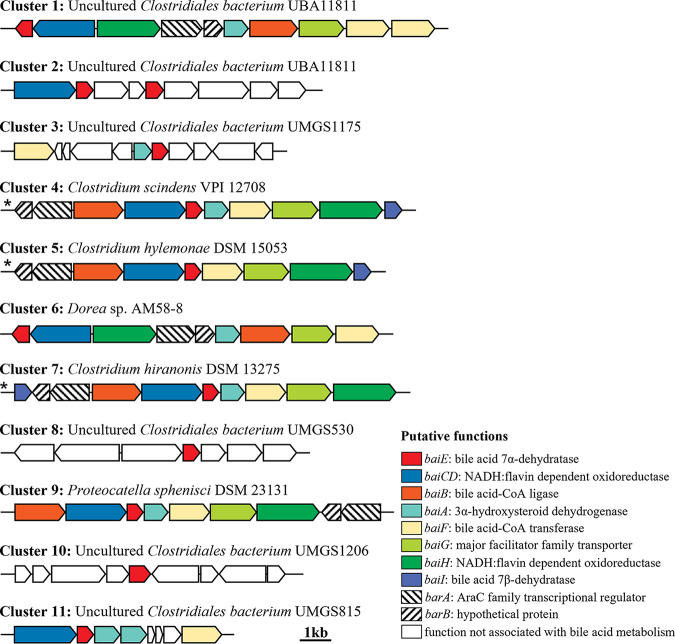
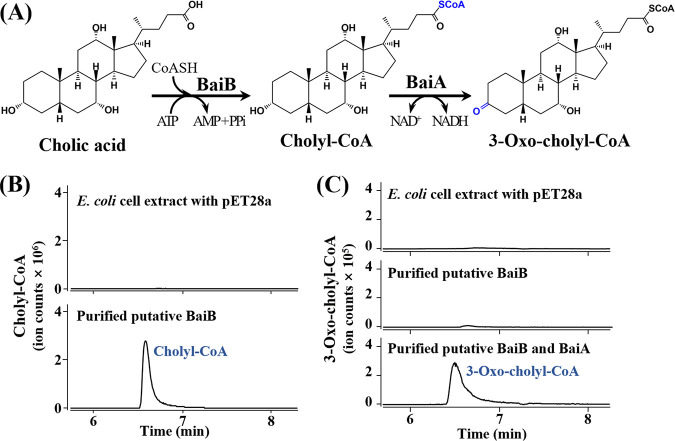
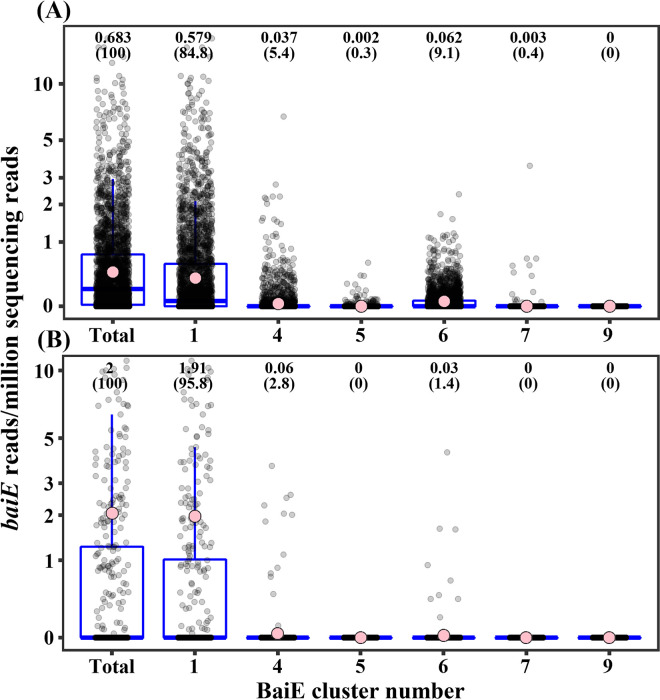
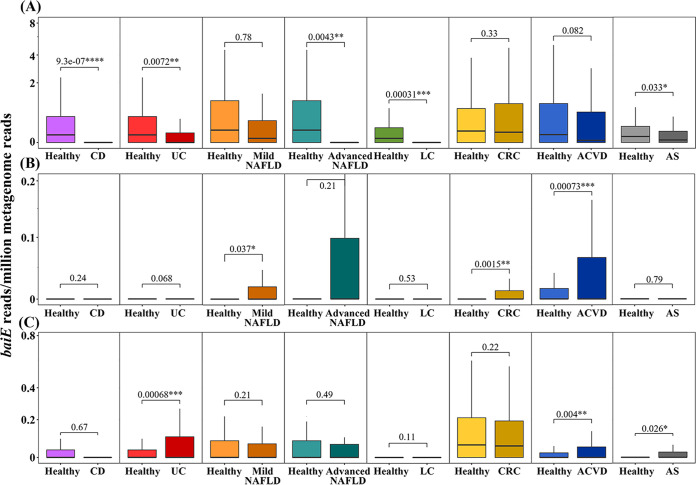
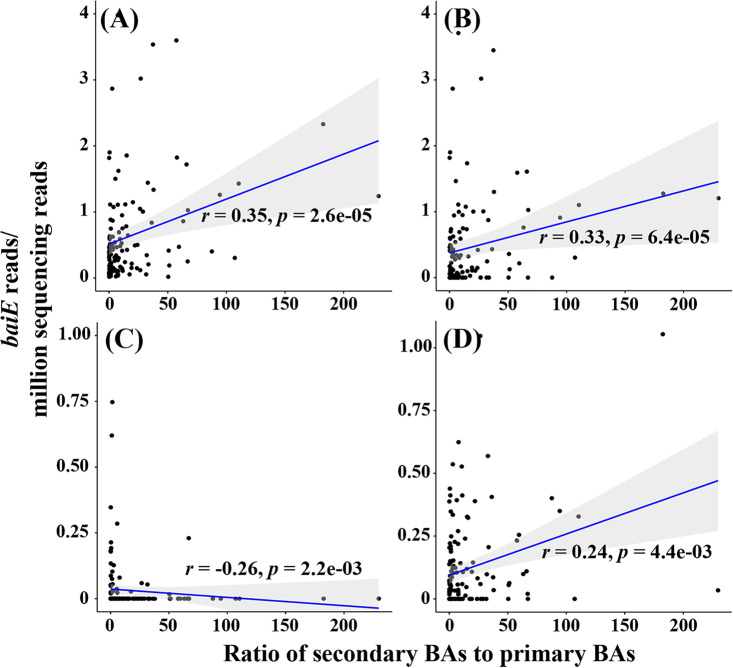
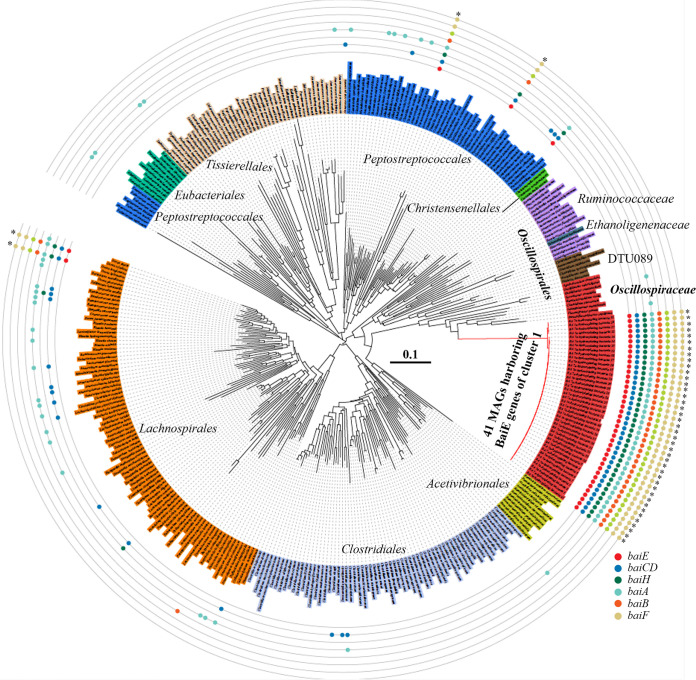
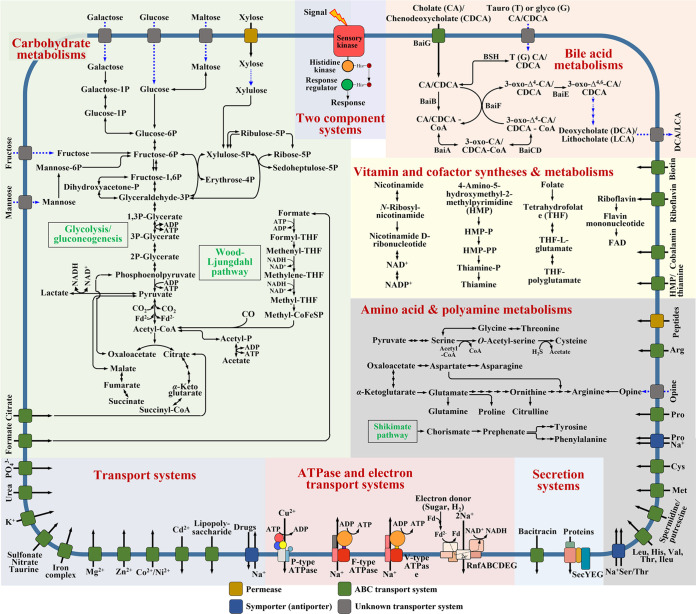
The metabolism of bile acids (BAs) by gut bacteria plays an important role in human health. This study identified and characterized 7α-dehydroxylating bacteria, which are majorly responsible for converting primary BAs to secondary BAs, in the human gut and investigated their association with human disease. Six 7α-dehydratase (BaiE) clusters were identified from human gut metagenomes through sequence similarity network and genome neighborhood network analyses. Abundance analyses of gut metagenomes and metatranscriptomes identified a cluster of bacteria (cluster 1) harboring baiE genes that may be key 7α-dehydroxylating bacteria in the human gut. The baiE gene abundance of cluster 1 was significantly and positively correlated with the ratio of secondary BAs to primary BAs. Furthermore, the baiE gene abundances of cluster 1 were significantly negatively correlated with inflammatory bowel disease, including Crohn's disease and ulcerative colitis, as well as advanced nonalcoholic fatty liver disease, liver cirrhosis, and ankylosing spondylitis. Phylogenetic and metagenome-assembled genome analyses showed that the 7α-dehydroxylating bacterial clade of cluster 1 was affiliated with the family Oscillospiraceae and may demonstrate efficient BA dehydroxylation ability by harboring both a complete bai operon, for proteins which produce secondary BAs from primary BAs, and a gene for bile salt hydrolase, which deconjugates BAs, in the human gut. IMPORTANCE In this study, we identified a key 7α-dehydroxylating bacterial group predicted to be largely responsible for converting primary bile acids (BAs) to secondary BAs in the human gut through sequence similarity network, genome neighborhood network, and gene abundance analyses using human gut metagenomes. The key bacterial group was phylogenetically quite different from known 7α-dehydroxylating bacteria, and their abundance was highly correlated with the occurrence of diverse diseases associated with bile acid 7α-dehydroxylation. In addition, we characterized the metabolic features of the key bacterial group using their metagenome-assembled genomes. This approach is useful to identify and characterize key gut bacteria highly associated with human health and diseases.
Keywords: 7α-dehydratase; BaiE; IBD; bile acids; human gut microbiota; metabolic pathways; sequence similarity networks.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Metabolism of Oxo-Bile Acids and Characterization of Recombinant 12α-Hydroxysteroid Dehydrogenases from Bile Acid 7α-Dehydroxylating Human Gut Bacteria.Appl Environ Microbiol. 2018 May 1;84(10):e00235-18. doi: 10.1128/AEM.00235-18. Print 2018 May 15. Appl Environ Microbiol. 2018. PMID: 29549099 Free PMC article.
-
Identification and characterization of a bile acid 7alpha-dehydroxylation operon in Clostridium sp. strain TO-931, a highly active 7alpha-dehydroxylating strain isolated from human feces.Appl Environ Microbiol. 2000 Mar;66(3):1107-13. doi: 10.1128/AEM.66.3.1107-1113.2000. Appl Environ Microbiol. 2000. PMID: 10698778 Free PMC article.
-
Isolation and characterization of a bile acid inducible 7alpha-dehydroxylating operon in Clostridium hylemonae TN271.Anaerobe. 2010 Apr;16(2):137-46. doi: 10.1016/j.anaerobe.2009.05.004. Epub 2009 May 21. Anaerobe. 2010. PMID: 19464381 Free PMC article.
-
Regulation of gut microbiota-bile acids axis by probiotics in inflammatory bowel disease.Front Immunol. 2022 Sep 23;13:974305. doi: 10.3389/fimmu.2022.974305. eCollection 2022. Front Immunol. 2022. PMID: 36211363 Free PMC article. Review.
-
Bile Acid and Gut Microbiota in Irritable Bowel Syndrome.J Neurogastroenterol Motil. 2022 Oct 30;28(4):549-561. doi: 10.5056/jnm22129. J Neurogastroenterol Motil. 2022. PMID: 36250362 Free PMC article. Review.
Cited by
-
Bile acids impact the microbiota, host, and C. difficile dynamics providing insight into mechanisms of efficacy of FMTs and microbiota-focused therapeutics.Gut Microbes. 2024 Jan-Dec;16(1):2393766. doi: 10.1080/19490976.2024.2393766. Epub 2024 Sep 3. Gut Microbes. 2024. PMID: 39224076 Free PMC article. Review.
-
Crosstalk between Gut Microbiota and Bile Acids in Cholestatic Liver Disease.Nutrients. 2023 May 22;15(10):2411. doi: 10.3390/nu15102411. Nutrients. 2023. PMID: 37242293 Free PMC article. Review.
-
Bile acids as a key target: traditional Chinese medicine for precision management of insulin resistance in type 2 diabetes mellitus through the gut microbiota-bile acids axis.Front Endocrinol (Lausanne). 2024 Dec 10;15:1481270. doi: 10.3389/fendo.2024.1481270. eCollection 2024. Front Endocrinol (Lausanne). 2024. PMID: 39720247 Free PMC article. Review.
-
Enhancing outcomes in medically inoperable early-stage NSCLC with gut-targeted antibiotics and stereotactic body radiotherapy: results from a randomized pilot study.J Immunother Cancer. 2025 Jul 10;13(7):e011356. doi: 10.1136/jitc-2024-011356. J Immunother Cancer. 2025. PMID: 40639850 Free PMC article. Clinical Trial.
-
Living Conditions Alter Ketogenic Diet-induced Metabolic Consequences in Mice through Modulating Gut Microbiota.Phenomics. 2024 Jun 6;4(4):313-326. doi: 10.1007/s43657-024-00161-1. eCollection 2024 Aug. Phenomics. 2024. PMID: 39583308 Free PMC article.
References
-
- Sato Y, Atarashi K, Plichta DR, Arai Y, Sasajima S, Kearney SM, Suda W, Takeshita K, Sasaki T, Okamoto S, Skelly AN, Okamura Y, Vlamakis H, Li Y, Tanoue T, Takei H, Nittono H, Narushima S, Irie J, Itoh H, Moriya K, Sugiura Y, Suematsu M, Moritoki N, Shibata S, Littman DR, Fischbach MA, Uwamino Y, Inoue T, Honda A, Hattori M, Murai T, Xavier RJ, Hirose N, Honda K. 2021. Novel bile acid biosynthetic pathways are enriched in the microbiome of centenarians. Nature 599:458–464. doi:10.1038/s41586-021-03832-5. - DOI - PubMed
-
- Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall HU, Bamberg K, Angelin B, Hyötyläinen T, Orešič M, Bäckhed F. 2013. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 17:225–235. doi:10.1016/j.cmet.2013.01.003. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
