

Molecular Pathophysiological Mechanisms in Huntington's Disease
- PMID: 35740453
- PMCID: PMC9219859
- DOI: 10.3390/biomedicines10061432
Molecular Pathophysiological Mechanisms in Huntington's Disease
Abstract
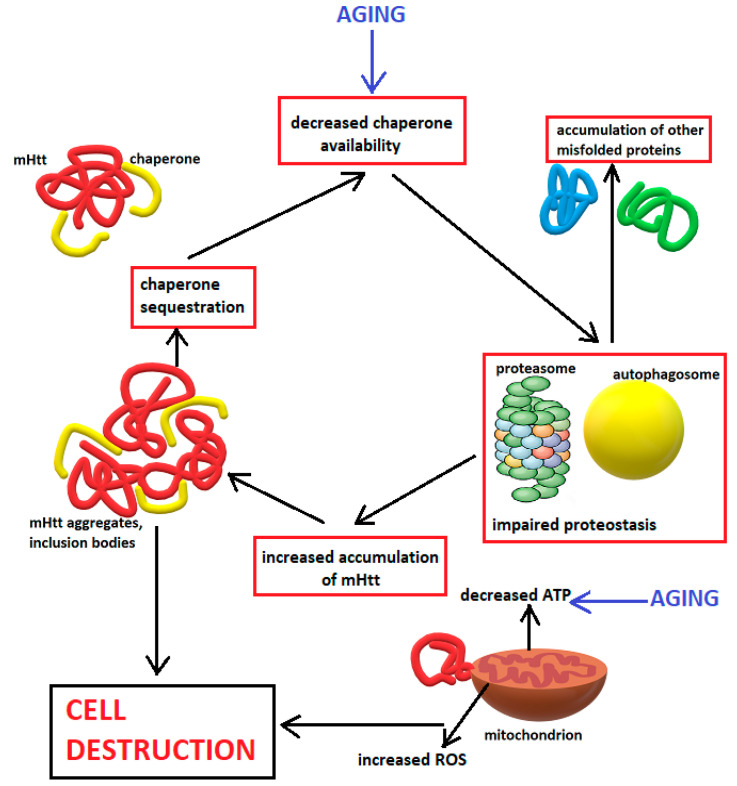
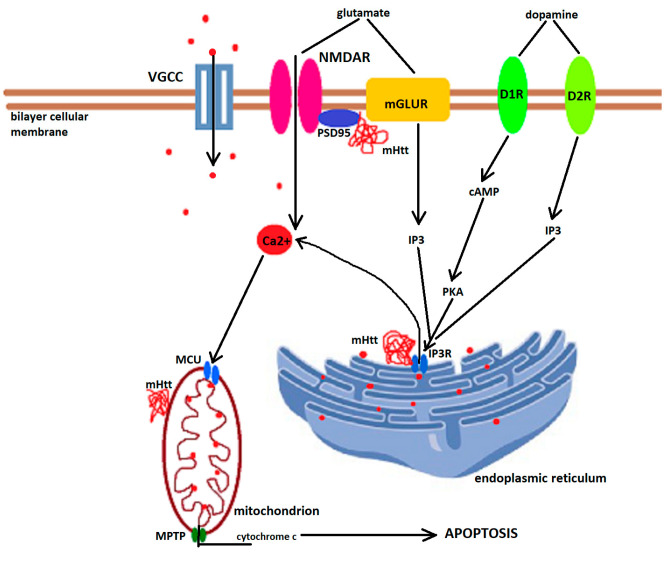
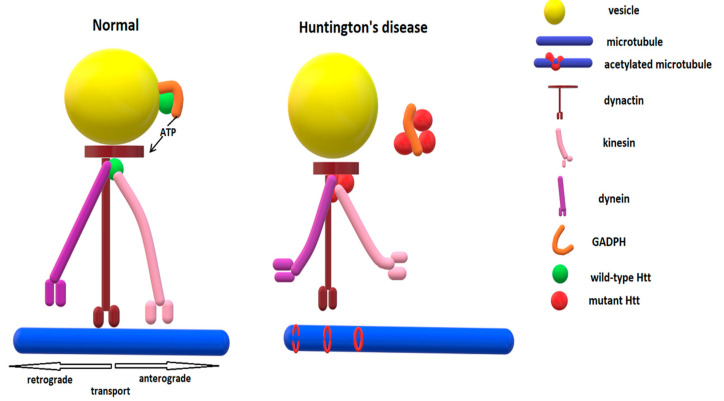
Huntington's disease is an inherited neurodegenerative disease described 150 years ago by George Huntington. The genetic defect was identified in 1993 to be an expanded CAG repeat on exon 1 of the huntingtin gene located on chromosome 4. In the following almost 30 years, a considerable amount of research, using mainly animal models or in vitro experiments, has tried to unravel the complex molecular cascades through which the transcription of the mutant protein leads to neuronal loss, especially in the medium spiny neurons of the striatum, and identified excitotoxicity, transcriptional dysregulation, mitochondrial dysfunction, oxidative stress, impaired proteostasis, altered axonal trafficking and reduced availability of trophic factors to be crucial contributors. This review discusses the pathogenic cascades described in the literature through which mutant huntingtin leads to neuronal demise. However, due to the ubiquitous presence of huntingtin, astrocytes are also dysfunctional, and neuroinflammation may additionally contribute to Huntington's disease pathology. The quest for therapies to delay the onset and reduce the rate of Huntington's disease progression is ongoing, but is based on findings from basic research.
Keywords: Huntington’s disease; animal models; brain-derived neurotrophic factor; excitotoxicity; mitochondrial dysfunction; mutant huntingtin; oxidative stress; proteostasis; transcriptional dysregulation.
Conflict of interest statement
The author declares no conflict of interest.
Figures
References
-
- Perandones C., Radrizzani M., Micheli F.E. Molecular Mechanisms Involved in the Pathogenesis of Huntington’s Disease. In: Visser T.J., editor. Huntington’s Disease: Etiology and Symptoms, Diagnosis and Treatment. Nova Science Publishers, Inc.; New York, NY, USA: 2010. pp. 1–37.
Publication types
LinkOut - more resources
Full Text Sources
