

Synaptic Dysfunction by Mutations in GRIN2B: Influence of Triheteromeric NMDA Receptors on Gain-of-Function and Loss-of-Function Mutant Classification
- PMID: 35741674
- PMCID: PMC9221112
- DOI: 10.3390/brainsci12060789
Synaptic Dysfunction by Mutations in GRIN2B: Influence of Triheteromeric NMDA Receptors on Gain-of-Function and Loss-of-Function Mutant Classification
Abstract
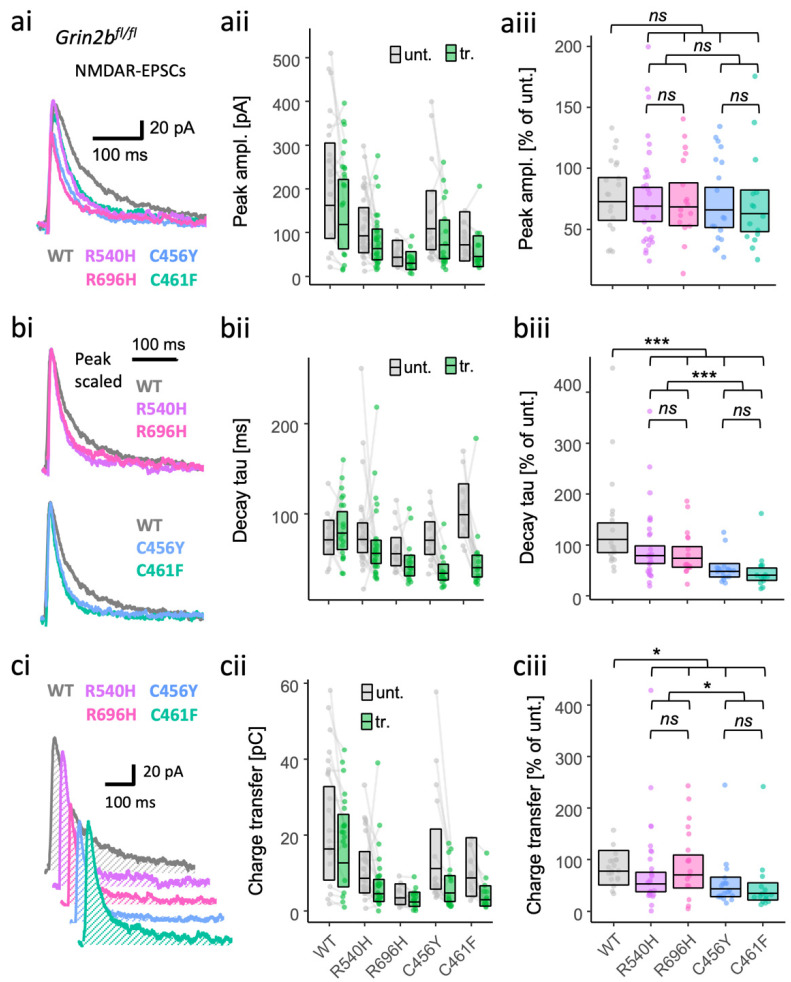
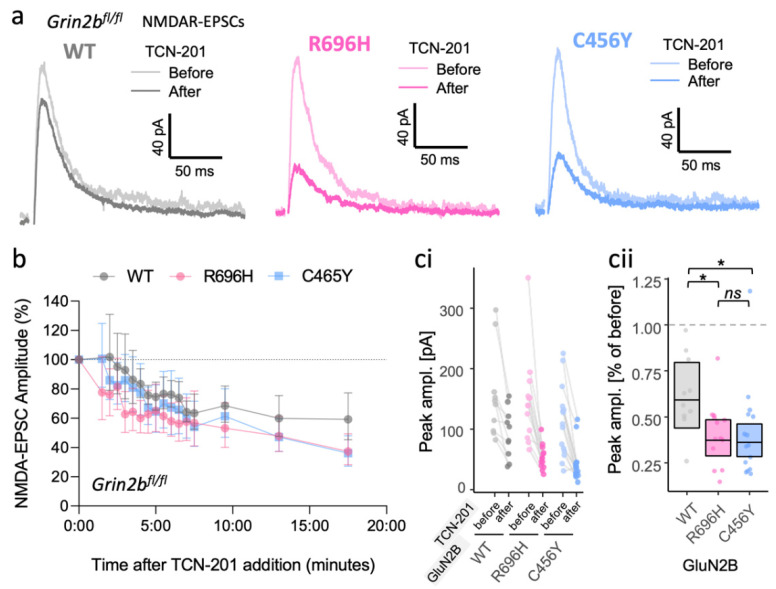
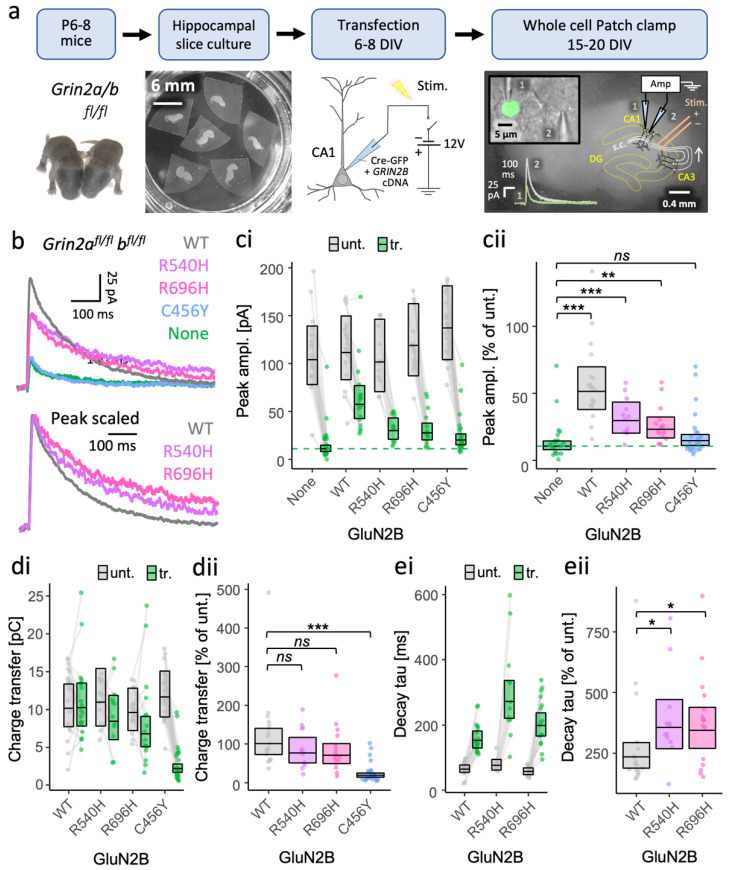
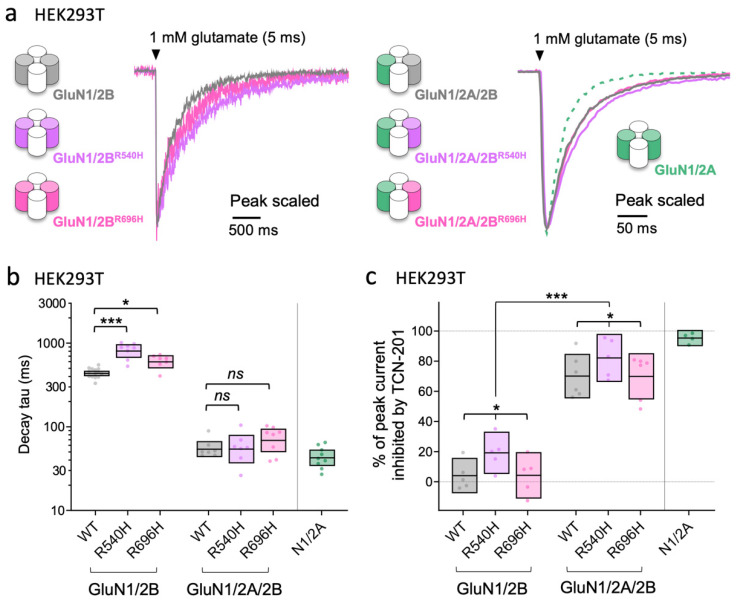
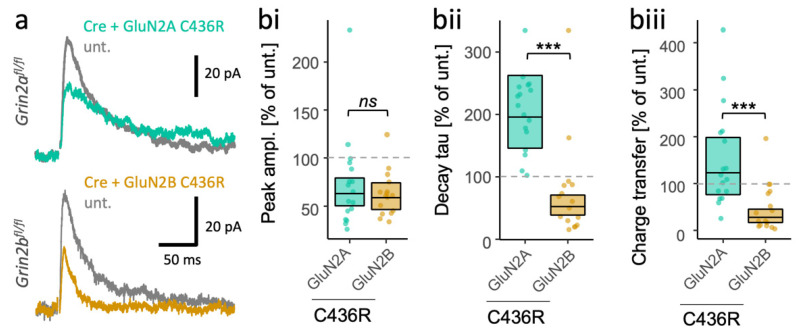
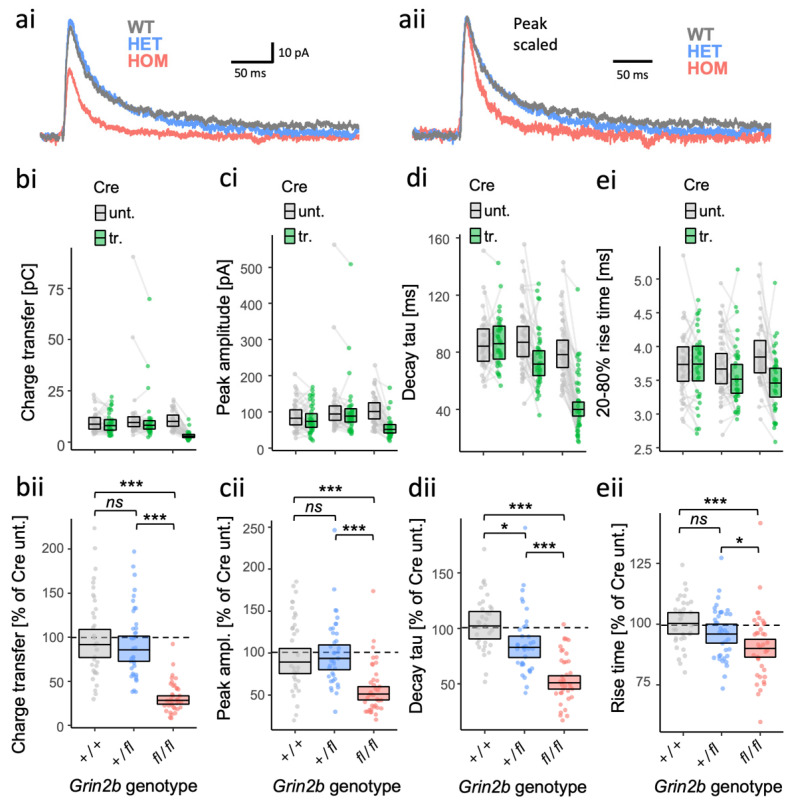
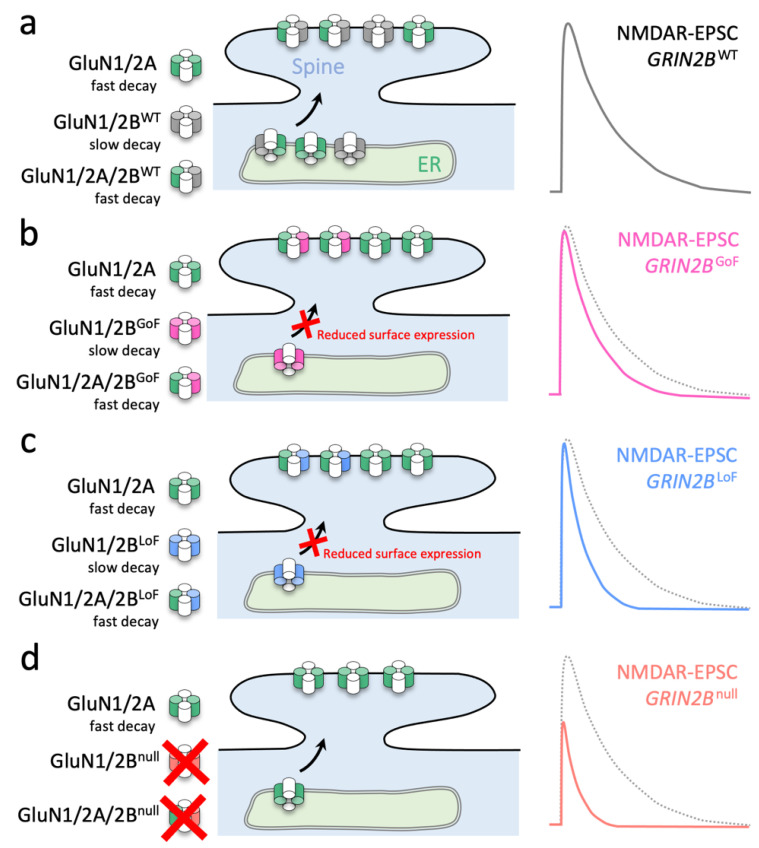
GRIN2B mutations are rare but often associated with patients having severe neurodevelopmental disorders with varying range of symptoms such as intellectual disability, developmental delay and epilepsy. Patient symptoms likely arise from mutations disturbing the role that the encoded NMDA receptor subunit, GluN2B, plays at neuronal connections in the developing nervous system. In this study, we investigated the cell-autonomous effects of putative gain- (GoF) and loss-of-function (LoF) missense GRIN2B mutations on excitatory synapses onto CA1 pyramidal neurons in organotypic hippocampal slices. In the absence of both native GluN2A and GluN2B subunits, functional incorporation into synaptic NMDA receptors was attenuated for GoF mutants, or almost eliminated for LoF GluN2B mutants. NMDA-receptor-mediated excitatory postsynaptic currents (NMDA-EPSCs) from synaptic GoF GluN1/2B receptors had prolonged decays consistent with their functional classification. Nonetheless, in the presence of native GluN2A, molecular replacement of native GluN2B with GoF and LoF GluN2B mutants all led to similar functional incorporation into synaptic receptors, more rapidly decaying NMDA-EPSCs and greater inhibition by TCN-201, a selective antagonist for GluN2A-containing NMDA receptors. Mechanistic insight was gained from experiments in HEK293T cells, which revealed that GluN2B GoF mutants slowed deactivation in diheteromeric GluN1/2B, but not triheteromeric GluN1/2A/2B receptors. We also show that a disease-associated missense mutation, which severely affects surface expression, causes opposing effects on NMDA-EPSC decay and charge transfer when introduced into GluN2A or GluN2B. Finally, we show that having a single null Grin2b allele has only a modest effect on NMDA-EPSC decay kinetics. Our results demonstrate that functional incorporation of GoF and LoF GluN2B mutants into synaptic receptors and the effects on EPSC decay times are highly dependent on the presence of triheteromeric GluN1/2A/2B NMDA receptors, thereby influencing the functional classification of NMDA receptor variants as GoF or LoF mutations. These findings highlight the complexity of interpreting effects of disease-causing NMDA receptor missense mutations in the context of neuronal function.
Keywords: central nervous system; de novo mutations; electrophysiology; ionotropic glutamate receptors; synaptic transmission.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures
References
-
- Hansen K.B., Wollmuth L.P., Bowie D., Furukawa H., Menniti F.S., Sobolevsky A.I., Swanson G.T., Swanger S.A., Greger I.H., Nakagawa T., et al. Structure, Function, and Pharmacology of Glutamate Receptor Ion Channels. Pharmacol. Rev. 2021;73:298–487. doi: 10.1124/pharmrev.120.000131. - DOI - PMC - PubMed
-
- Kew J.N., Koester A., Moreau J.L., Jenck F., Ouagazzal A.M., Mutel V., Richards J.G., Trube G., Fischer G., Montkowski A., et al. Functional Consequences of Reduction in NMDA Receptor Glycine Affinity in Mice Carrying Targeted Point Mutations in the Glycine Binding Site. J. Neurosci. 2000;20:4037–4049. doi: 10.1523/JNEUROSCI.20-11-04037.2000. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
