

The RelB-BLNK Axis Determines Cellular Response to a Novel Redox-Active Agent Betamethasone during Radiation Therapy in Prostate Cancer
- PMID: 35742868
- PMCID: PMC9223669
- DOI: 10.3390/ijms23126409
The RelB-BLNK Axis Determines Cellular Response to a Novel Redox-Active Agent Betamethasone during Radiation Therapy in Prostate Cancer
Abstract
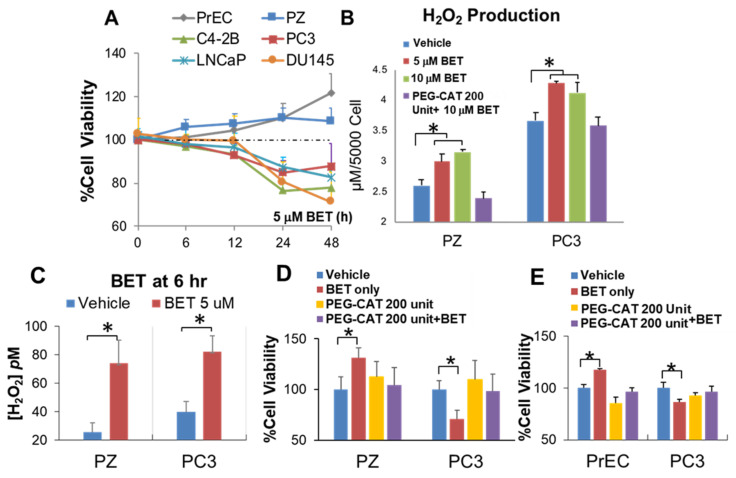
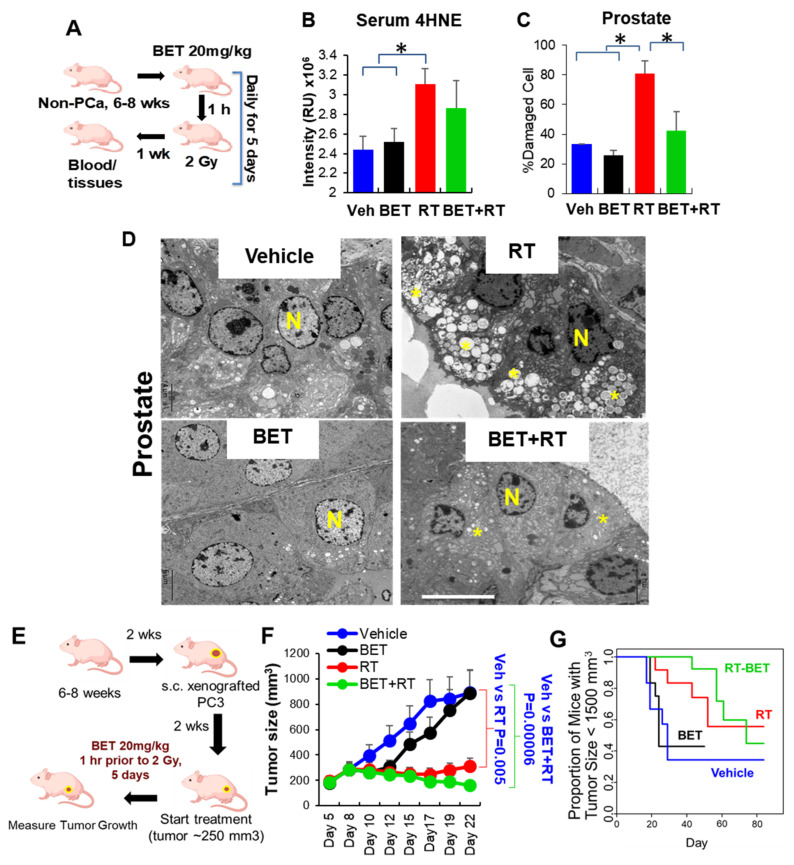
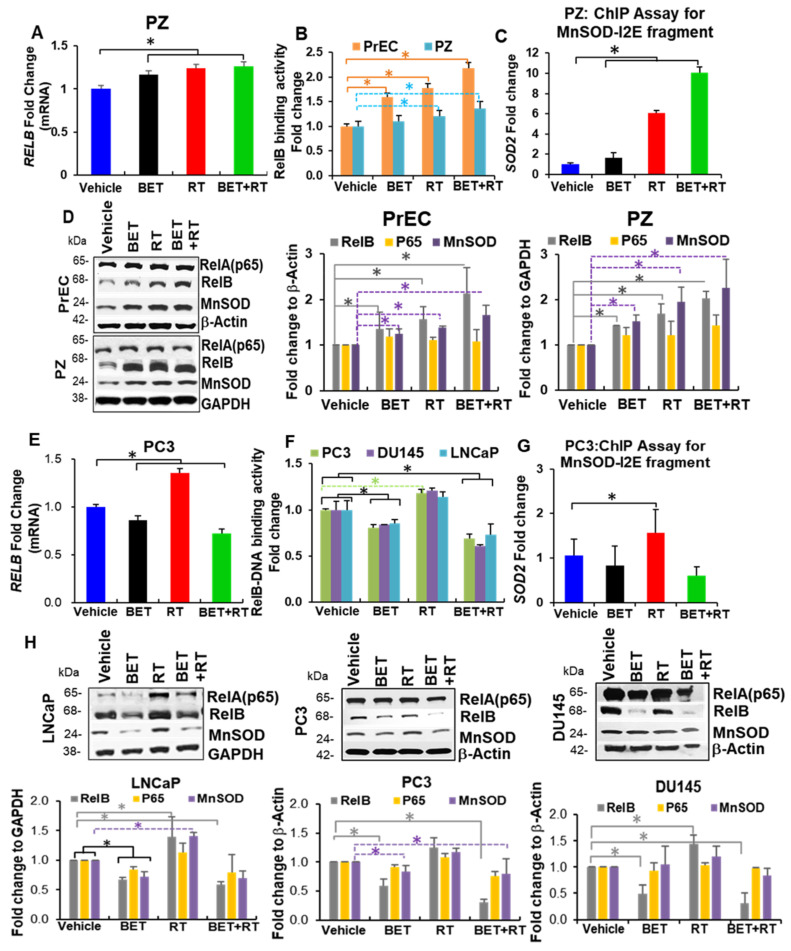
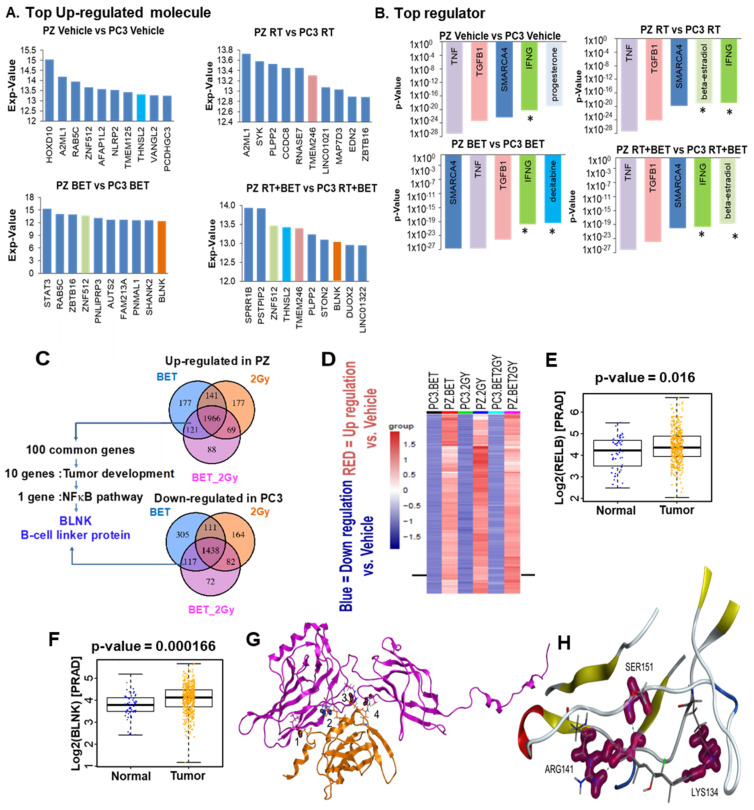
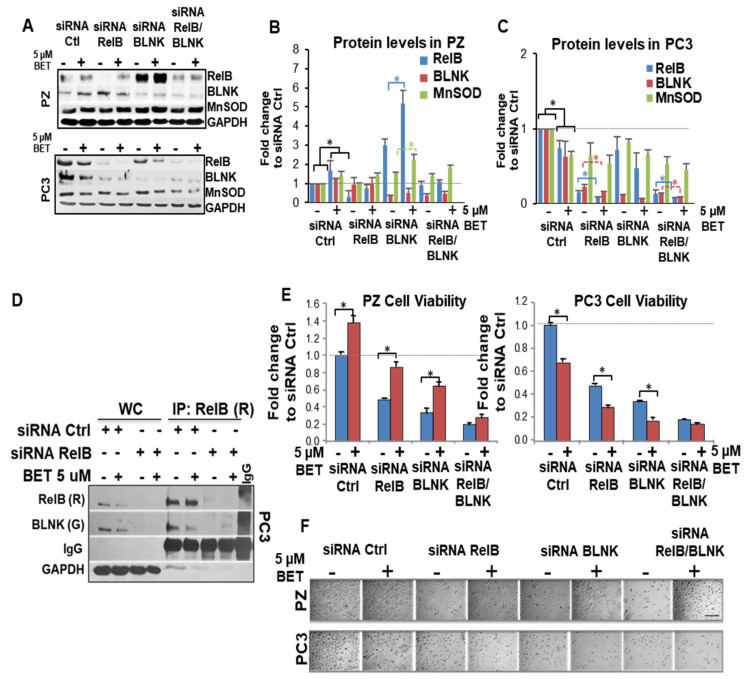
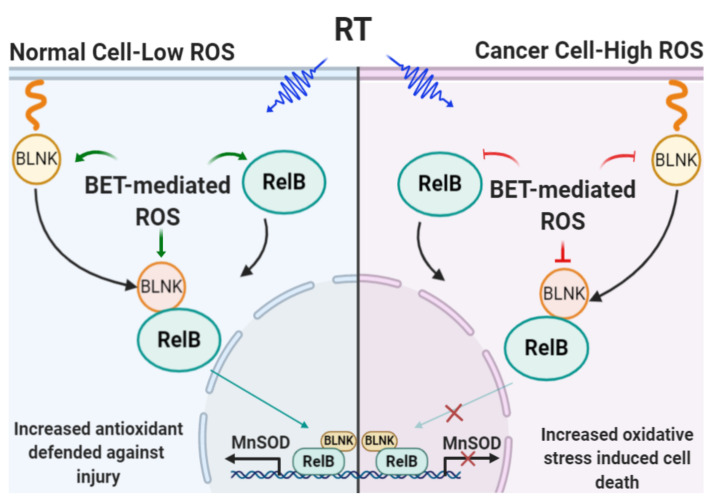
Aberrant levels of reactive oxygen species (ROS) are potential mechanisms that contribute to both cancer therapy efficacy and the side effects of cancer treatment. Upregulation of the non-canonical redox-sensitive NF-kB family member, RelB, confers radioresistance in prostate cancer (PCa). We screened FDA-approved compounds and identified betamethasone (BET) as a drug that increases hydrogen peroxide levels in vitro and protects non-PCa tissues/cells while also enhancing radiation killing of PCa tissues/cells, both in vitro and in vivo. Significantly, BET increases ROS levels and exerts different effects on RelB expression in normal cells and PCa cells. BET induces protein expression of RelB and RelB target genes, including the primary antioxidant enzyme, manganese superoxide dismutase (MnSOD), in normal cells, while it suppresses protein expression of RelB and MnSOD in LNCaP cells and PC3 cells. RNA sequencing analysis identifies B-cell linker protein (BLNK) as a novel RelB complementary partner that BET differentially regulates in normal cells and PCa cells. RelB and BLNK are upregulated and correlate with the aggressiveness of PCa in human samples. The RelB-BLNK axis translocates to the nuclear compartment to activate MnSOD protein expression. BET promotes the RelB-BLNK axis in normal cells but suppresses the RelB-BLNK axis in PCa cells. Targeted disruptions of RelB-BLNK expressions mitigate the radioprotective effect of BET on normal cells and the radiosensitizing effect of BET on PCa cells. Our study identified a novel RelB complementary partner and reveals a complex redox-mediated mechanism showing that the RelB-BLNK axis, at least in part, triggers differential responses to the redox-active agent BET by stimulating adaptive responses in normal cells but pushing PCa cells into oxidative stress overload.
Keywords: BLNK; RelB; betamethasone; radiation; redox state.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
