

Alisol B Alleviates Hepatocyte Lipid Accumulation and Lipotoxicity via Regulating RARα-PPARγ-CD36 Cascade and Attenuates Non-Alcoholic Steatohepatitis in Mice
- PMID: 35745142
- PMCID: PMC9231195
- DOI: 10.3390/nu14122411
Alisol B Alleviates Hepatocyte Lipid Accumulation and Lipotoxicity via Regulating RARα-PPARγ-CD36 Cascade and Attenuates Non-Alcoholic Steatohepatitis in Mice
Abstract
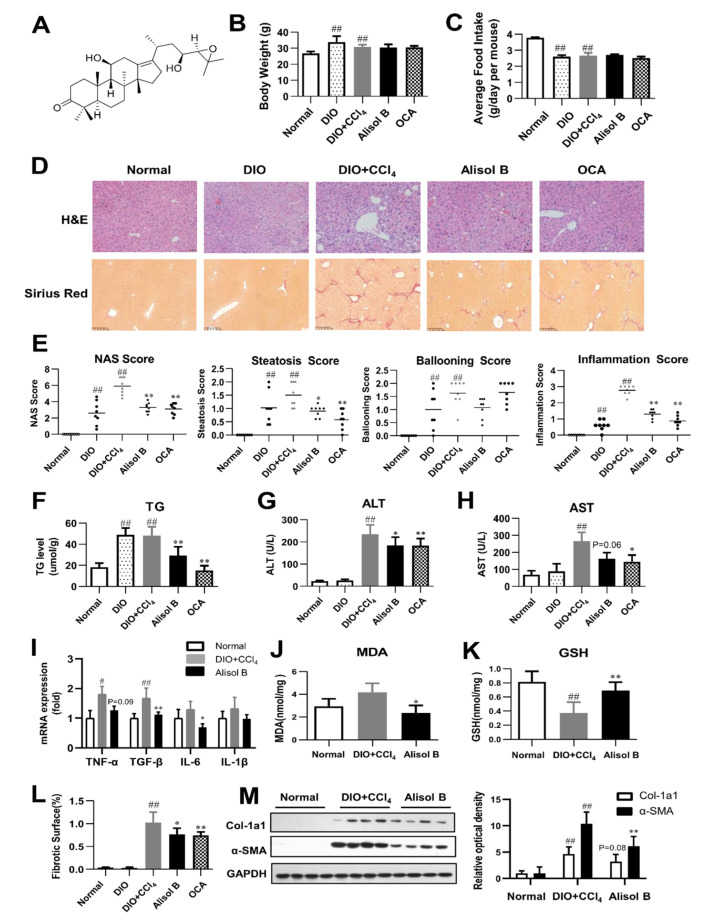
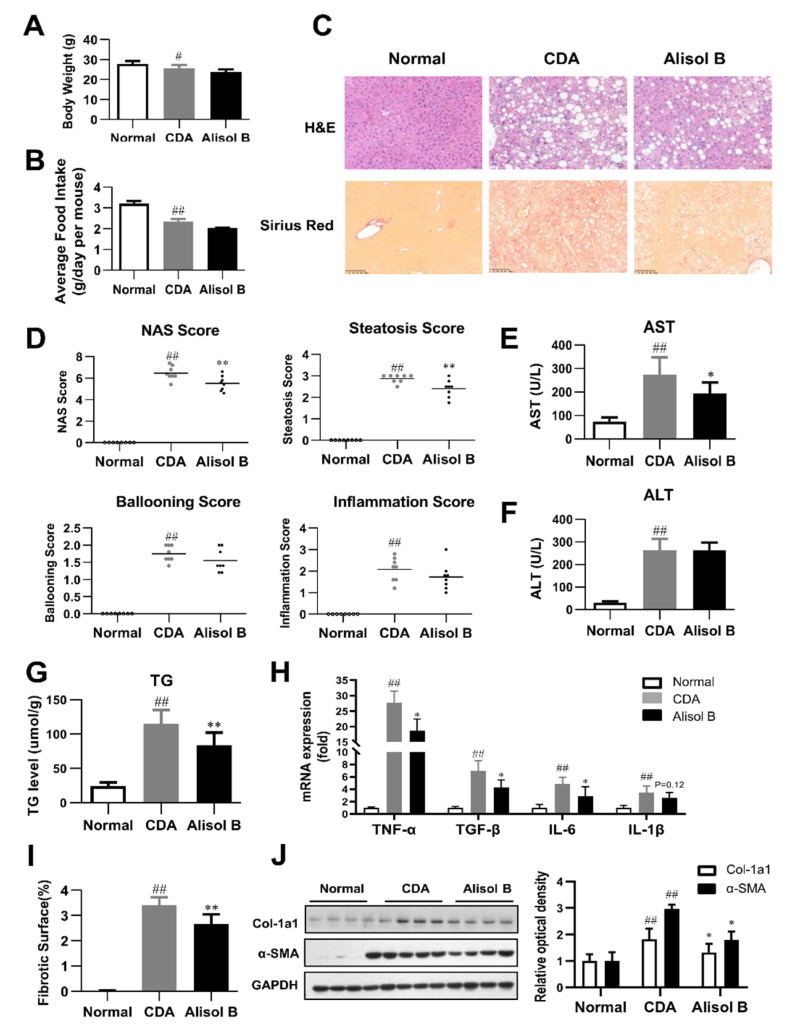
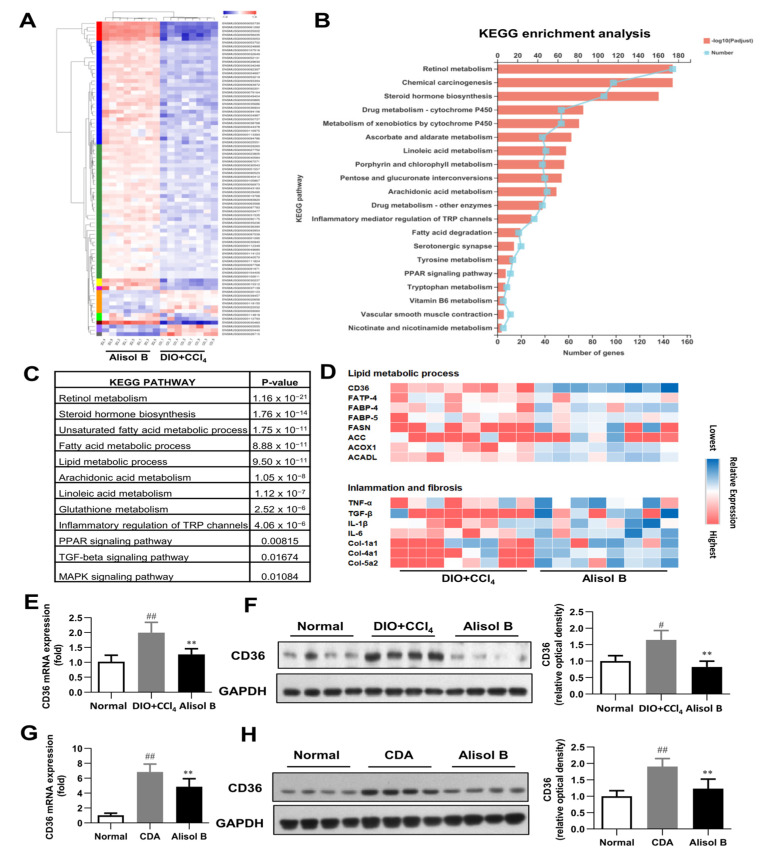
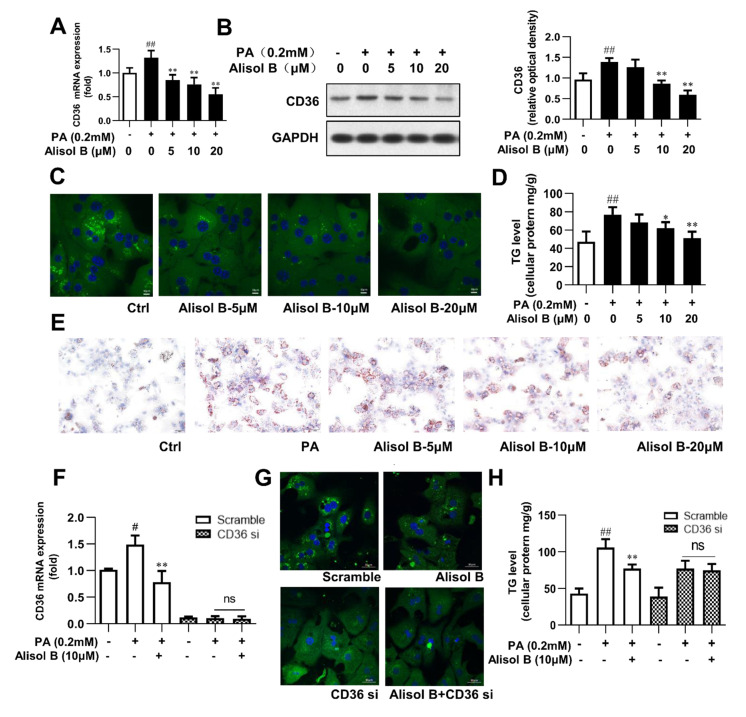
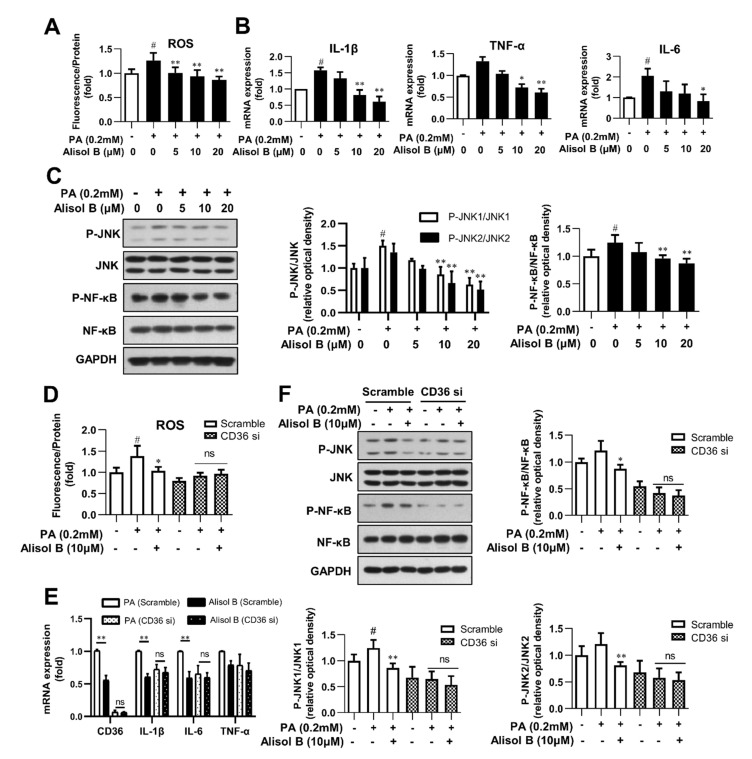
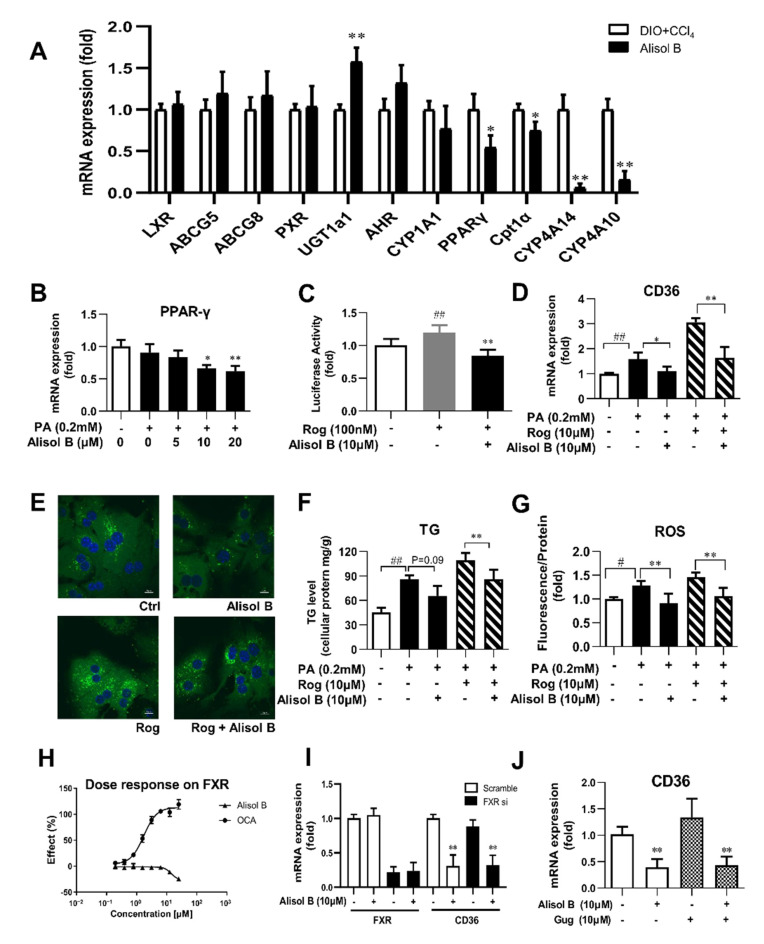
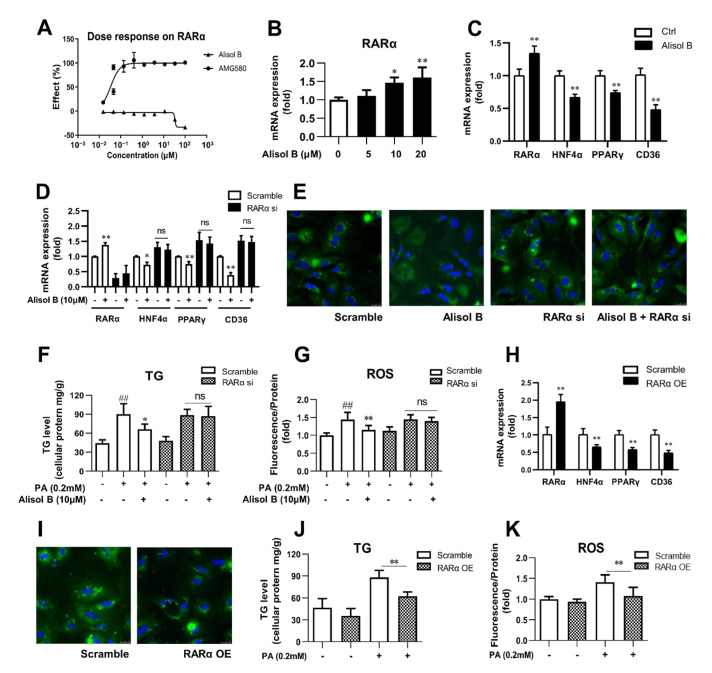
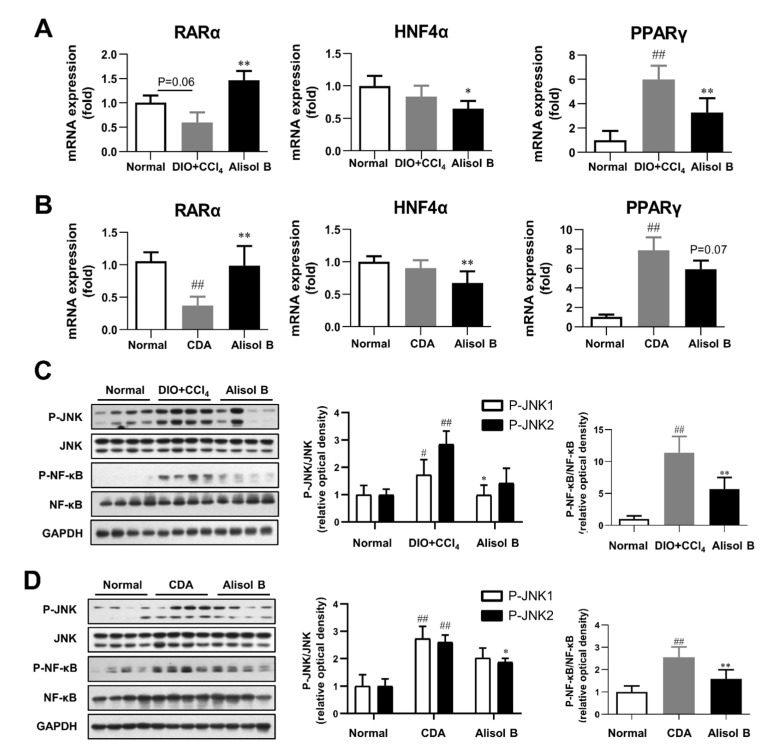
Non-alcoholic steatohepatitis (NASH) is a common chronic liver disease worldwide, with no effective therapies available. Discovering lead compounds from herb medicine might be a valuable strategy for the treatment of NASH. Here, we discovered Alisol B, a natural compound isolated from Alisma orientalis (Sam.), that attenuated hepatic steatosis, inflammation, and fibrosis in high-fat diet plus carbon tetrachloride (DIO+CCl4)-induced and choline-deficient and amino acid-defined (CDA)-diet-induced NASH mice. RNA-seq showed Alisol B significantly suppressed CD36 expression and regulated retinol metabolism in NASH mice. In mouse primary hepatocytes, Alisol B decreased palmitate-induced lipid accumulation and lipotoxicity, which were dependent on CD36 suppression. Further study revealed that Alisol B enhanced the gene expression of RARα with no direct RARα agonistic activity. The upregulation of RARα by Alisol B reduced HNF4α and PPARγ expression and further decreased CD36 expression. This effect was fully abrogated after RARα knockdown, suggesting Alisol B suppressed CD36 via regulating RARα-HNF4α-PPARγ cascade. Moreover, the hepatic gene expression of RARα was obviously decreased in murine NASH models, whereas Alisol B significantly increased RARα expression and decreased CD36 expression, along with the downregulation of HNF4α and PPARγ. Therefore, this study showed the unrecognized therapeutic effects of Alisol B against NASH with a novel mechanism by regulating RARα-PPARγ-CD36 cascade and highlighted Alisol B as a promising lead compound for the treatment of NASH.
Keywords: Alisol B; CD36; RARα; non-alcoholic steatohepatitis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
