

Discovery of Therapeutics Targeting Oxidative Stress in Autosomal Recessive Cerebellar Ataxia: A Systematic Review
- PMID: 35745683
- PMCID: PMC9228961
- DOI: 10.3390/ph15060764
Discovery of Therapeutics Targeting Oxidative Stress in Autosomal Recessive Cerebellar Ataxia: A Systematic Review
Abstract
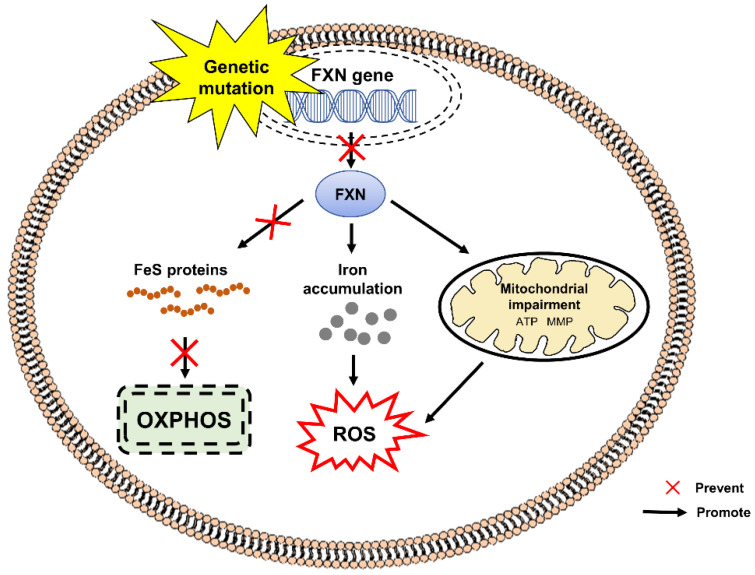
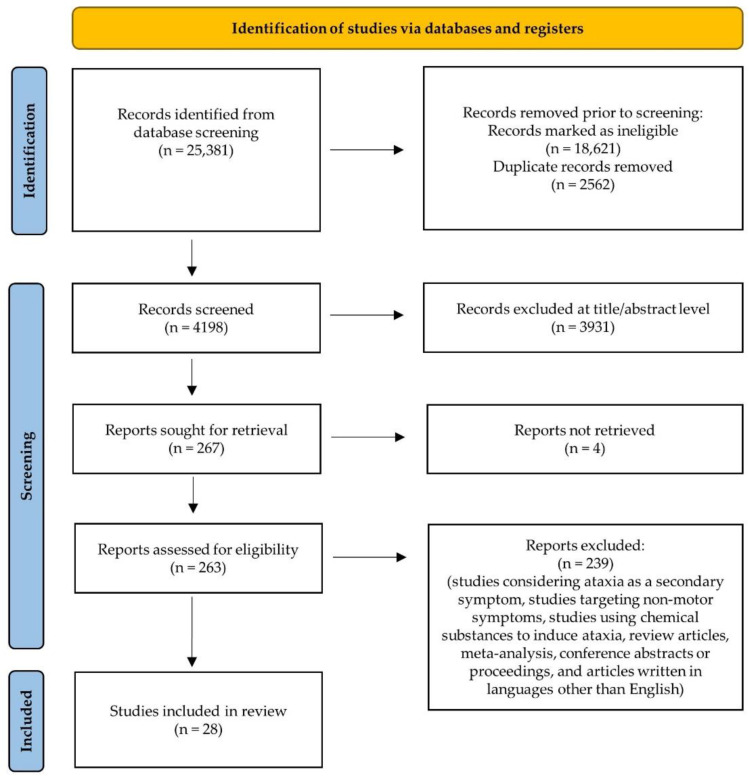
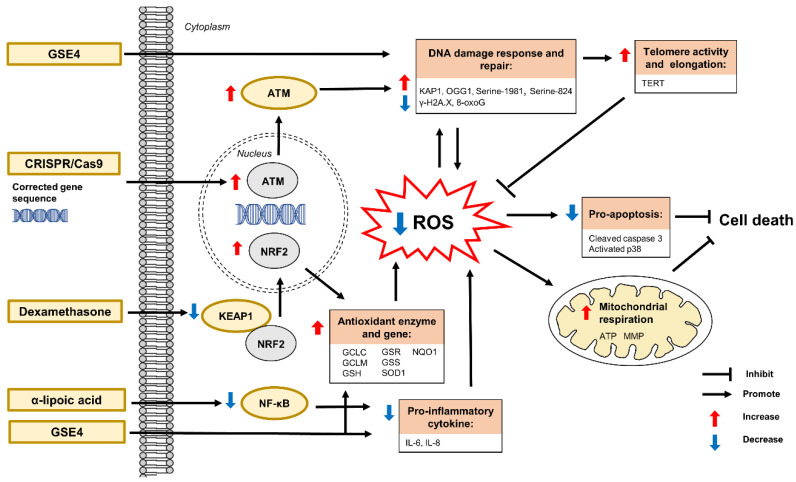
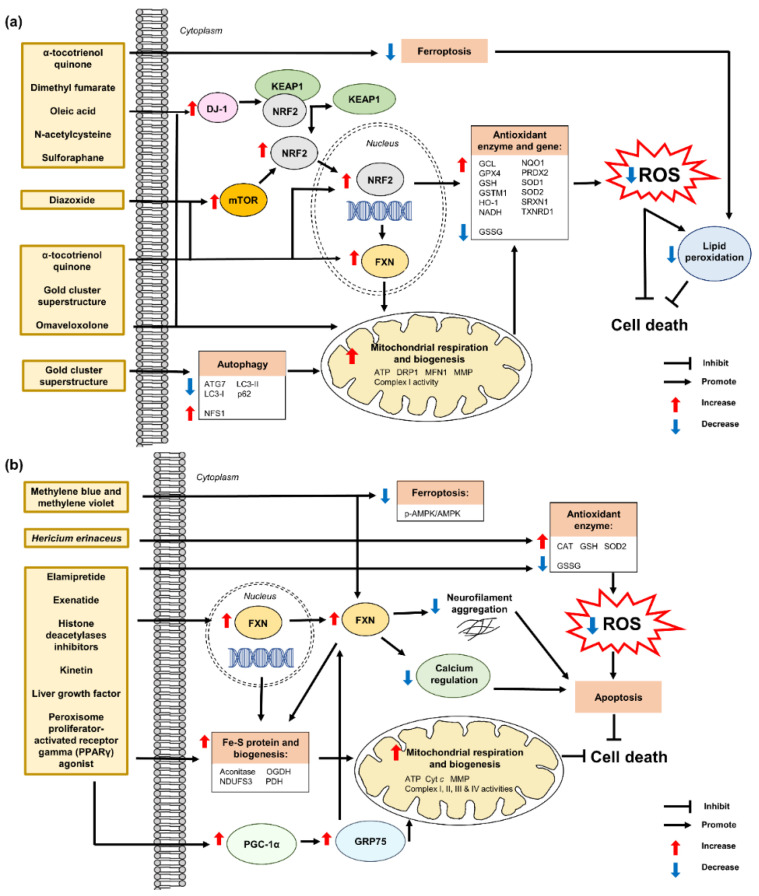
Autosomal recessive cerebellar ataxias (ARCAs) are a heterogeneous group of rare neurodegenerative inherited disorders. The resulting motor incoordination and progressive functional disabilities lead to reduced lifespan. There is currently no cure for ARCAs, likely attributed to the lack of understanding of the multifaceted roles of antioxidant defense and the underlying mechanisms. This systematic review aims to evaluate the extant literature on the current developments of therapeutic strategies that target oxidative stress for the management of ARCAs. We searched PubMed, Web of Science, and Science Direct Scopus for relevant peer-reviewed articles published from 1 January 2016 onwards. A total of 28 preclinical studies fulfilled the eligibility criteria for inclusion in this systematic review. We first evaluated the altered cellular processes, abnormal signaling cascades, and disrupted protein quality control underlying the pathogenesis of ARCA. We then examined the current potential therapeutic strategies for ARCAs, including aromatic, organic and pharmacological compounds, gene therapy, natural products, and nanotechnology, as well as their associated antioxidant pathways and modes of action. We then discussed their potential as antioxidant therapeutics for ARCAs, with the long-term view toward their possible translation to clinical practice. In conclusion, our current understanding is that these antioxidant therapies show promise in improving or halting the progression of ARCAs. Tailoring the therapies to specific disease stages could greatly facilitate the management of ARCAs.
Keywords: antioxidant pathway and therapy; autosomal recessive cerebellar ataxia; genetic mutation; oxidative stress; preclinical model; rare neurodegenerative disease.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures
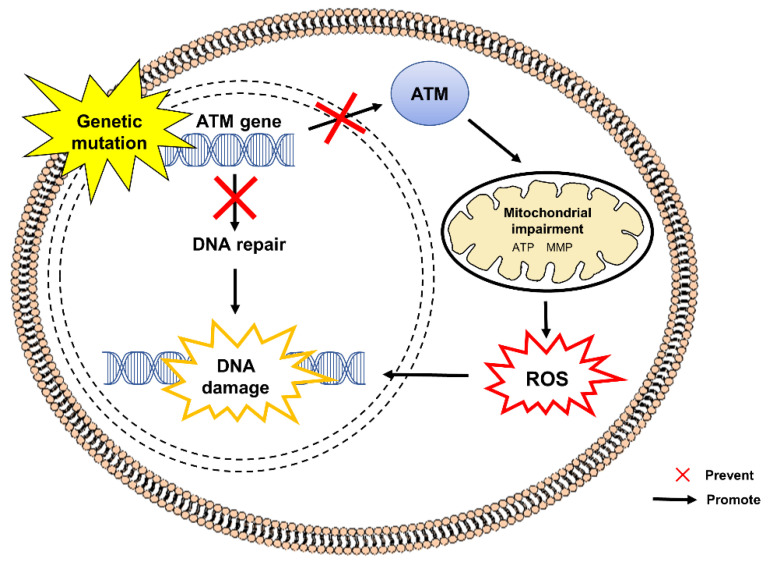
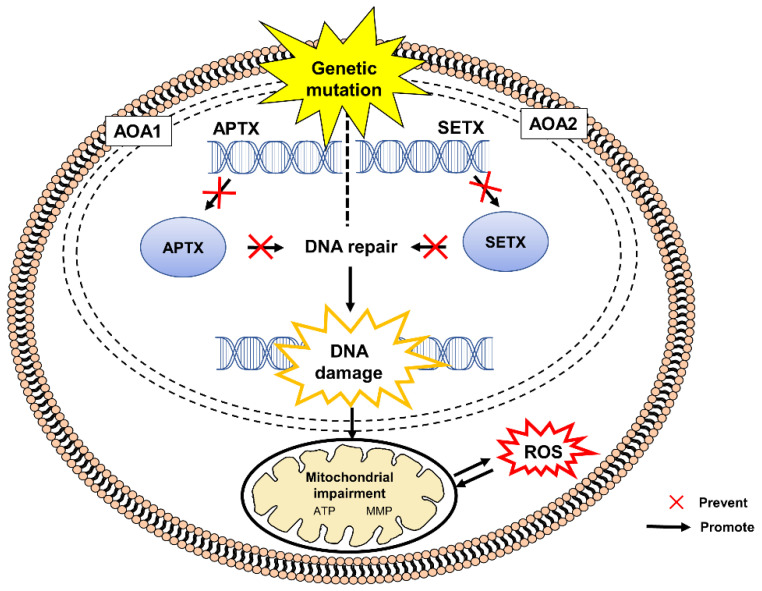
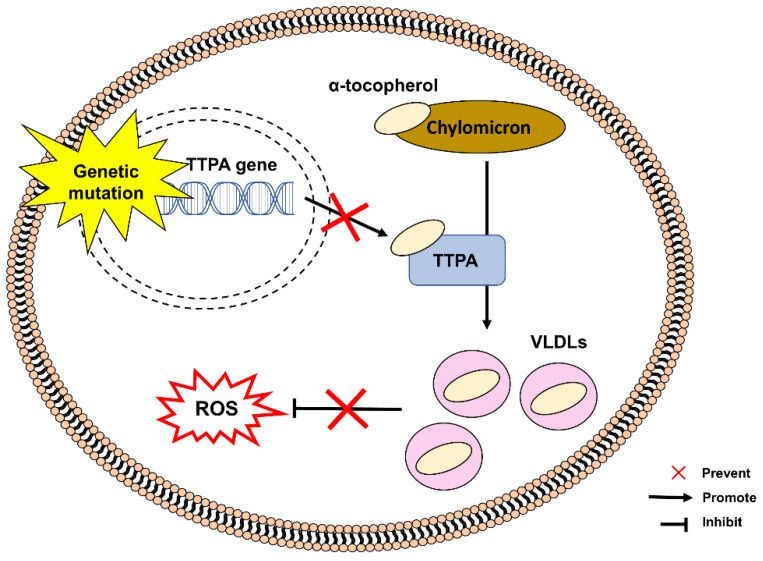
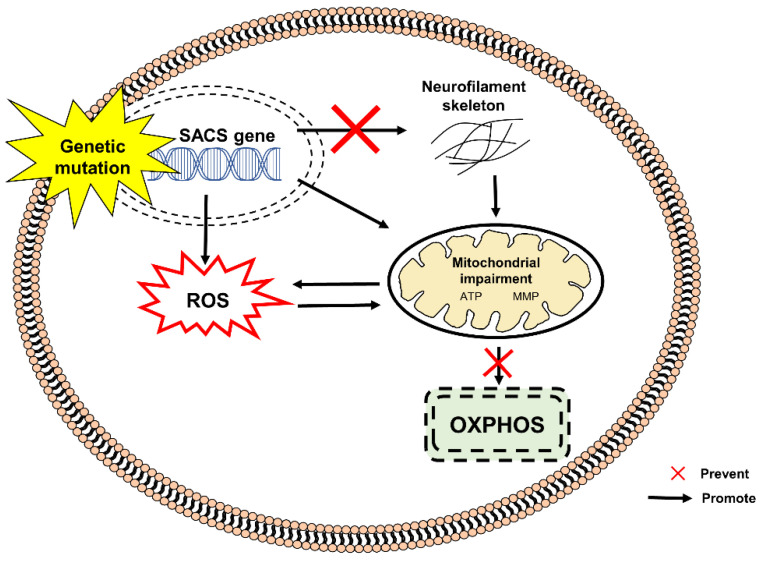
References
-
- Manto M.U., editor. Cerebellar Disorders: A Practical Approach to Diagnosis and Management. Cambridge University Press; Cambridge, UK: 2010. Autosomal recessive cerebellar ataxias; pp. 189–228. - DOI
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
