

The Process of Filopodia Induction during HPV Infection
- PMID: 35746622
- PMCID: PMC9231133
- DOI: 10.3390/v14061150
The Process of Filopodia Induction during HPV Infection
Abstract
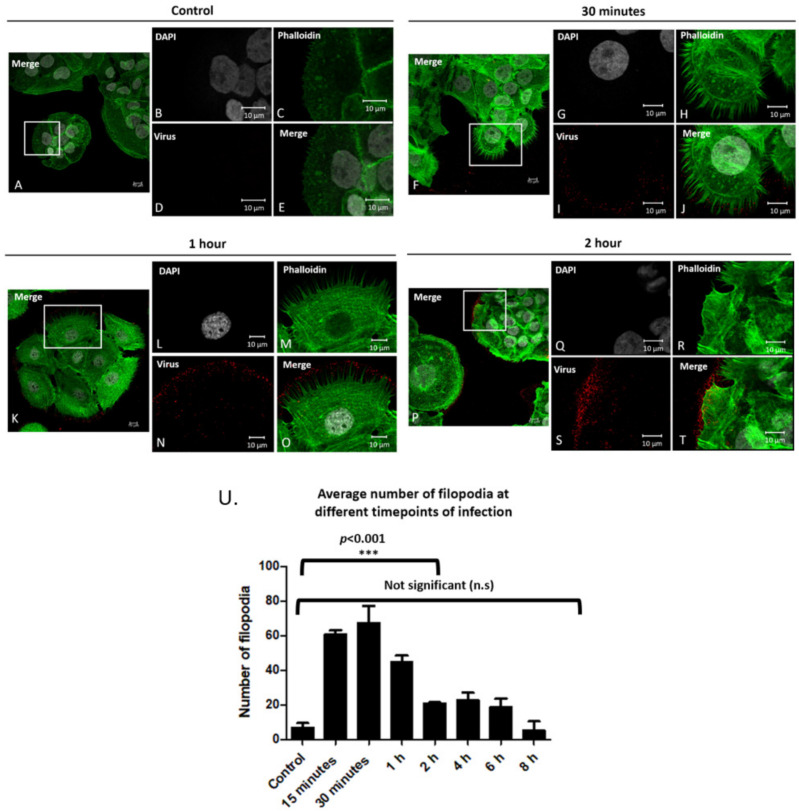
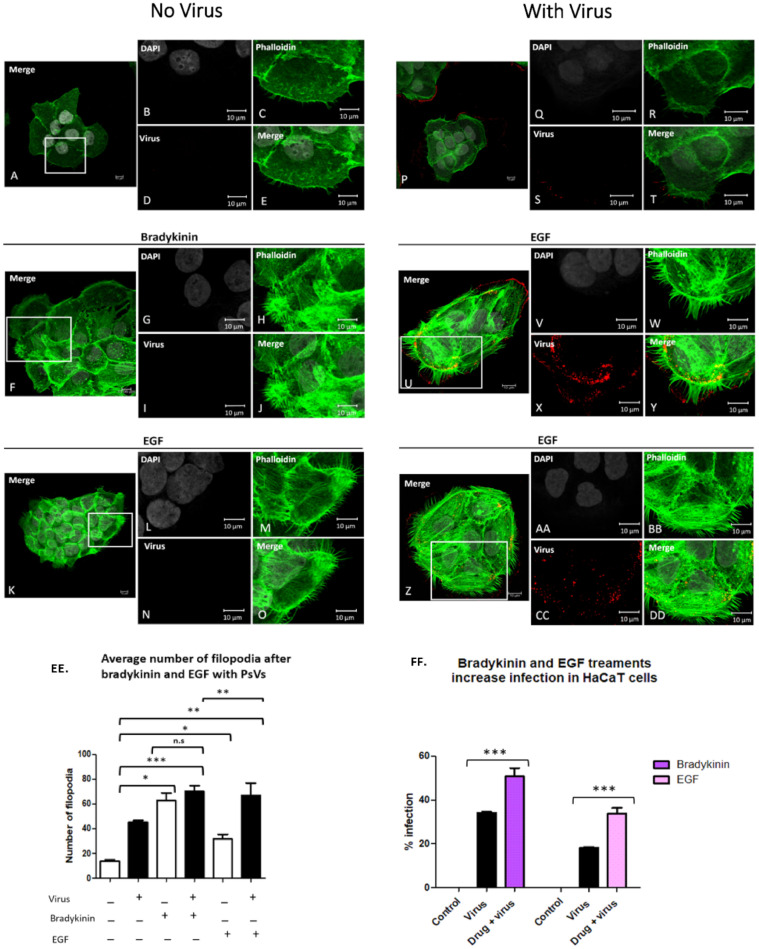
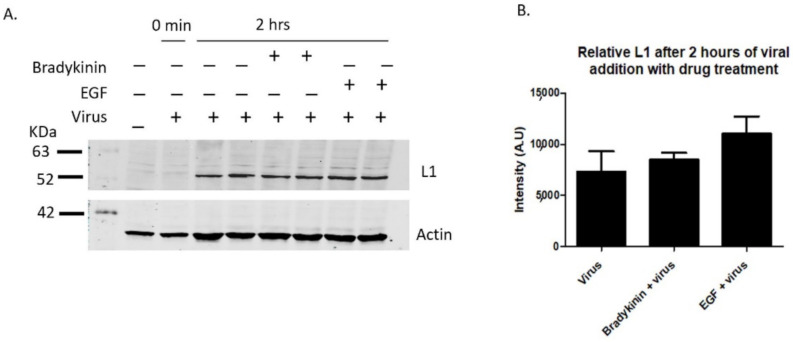
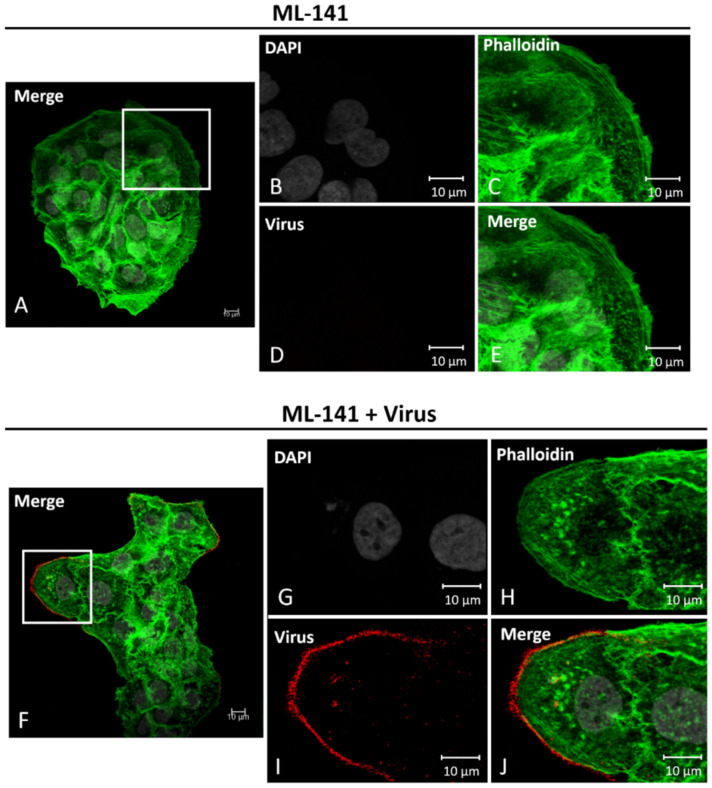
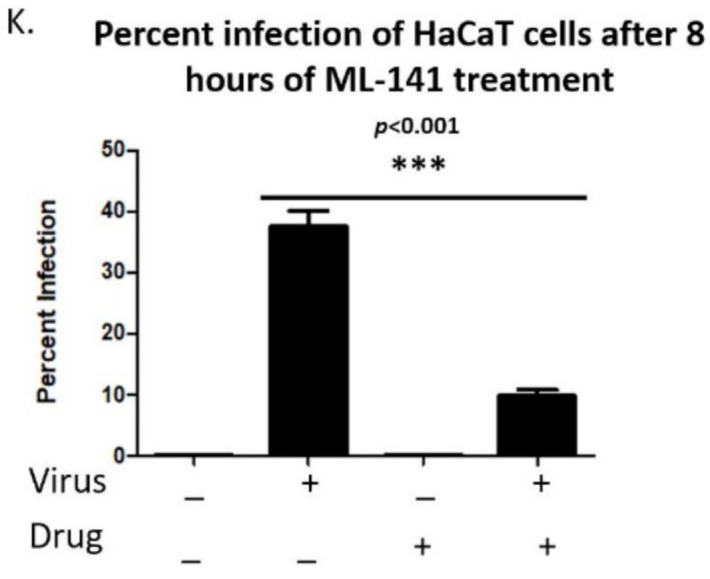
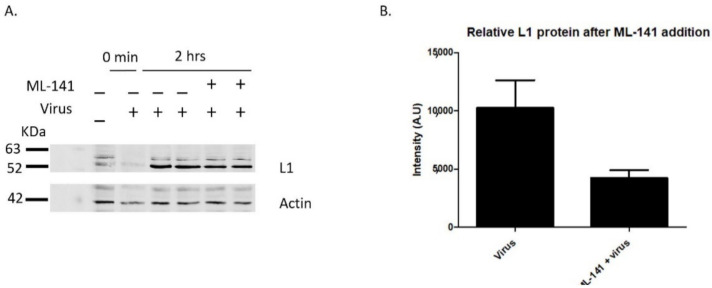
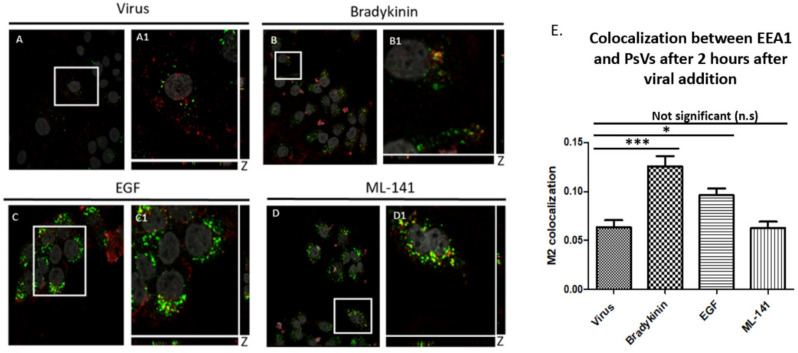
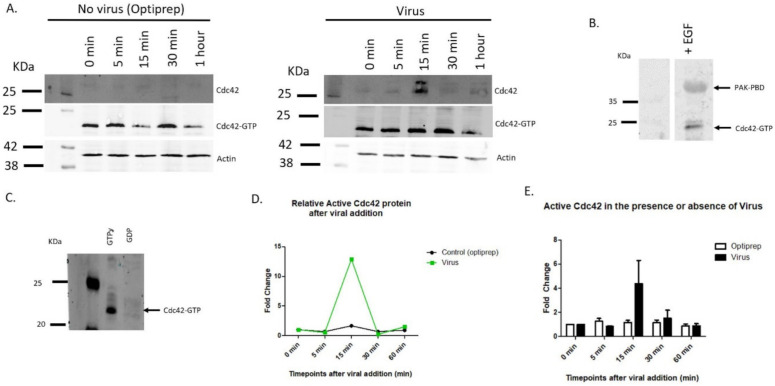
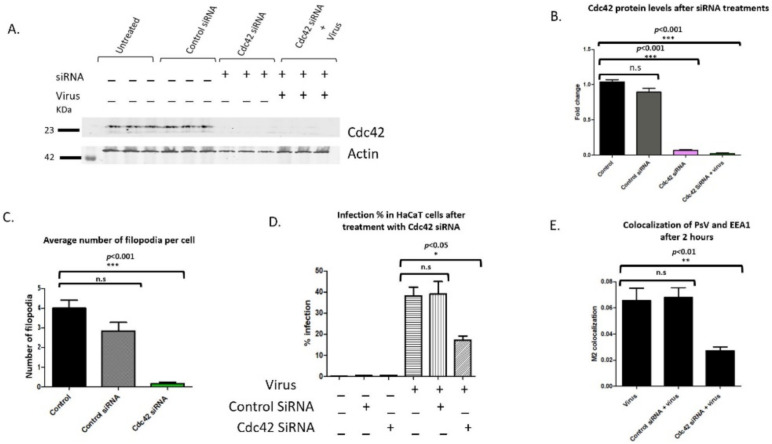
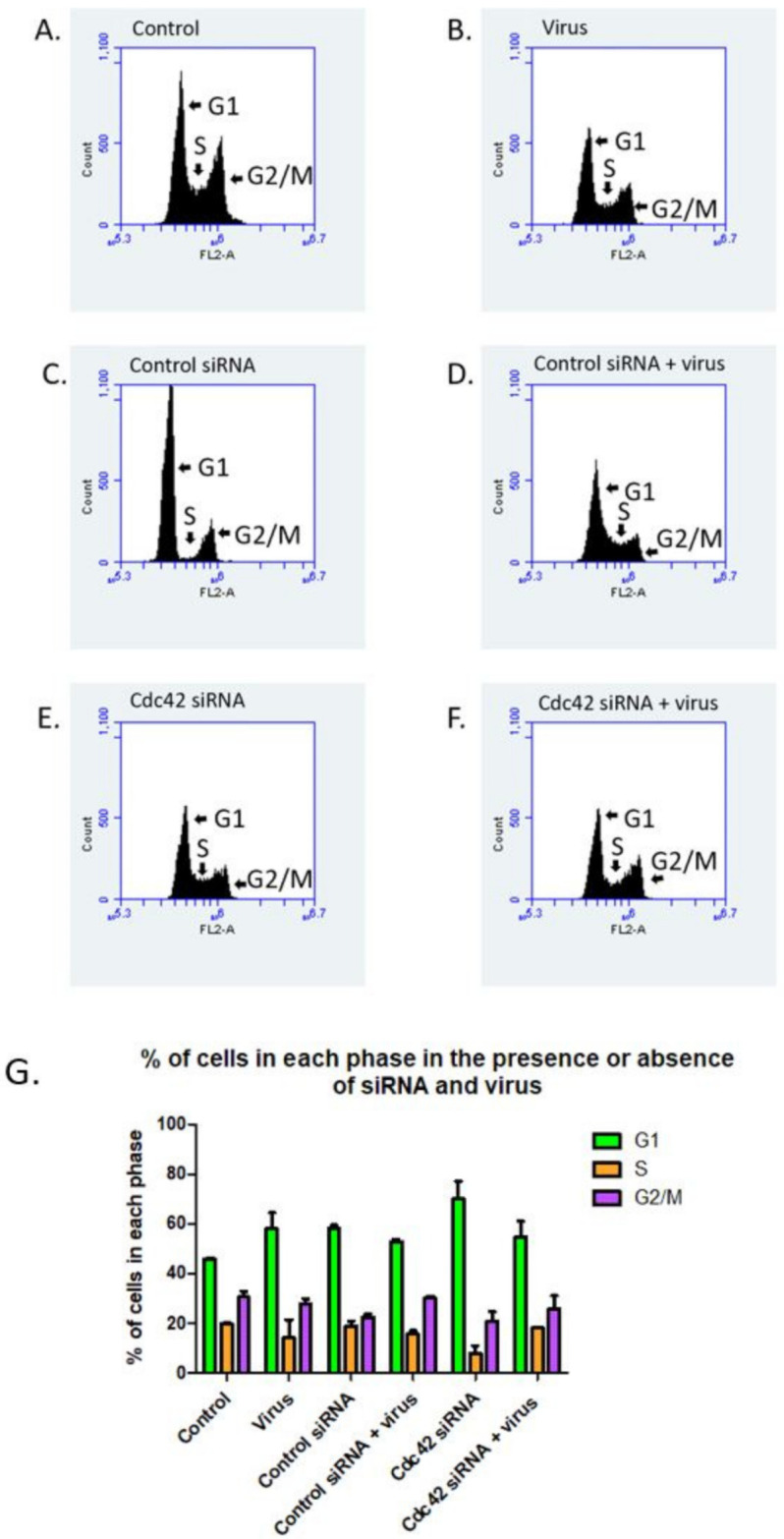
Human Papillomavirus 16 (HPV16) infects mucosal and epithelial cells and has been identified as a high-risk HPV type that is an etiologic agent of human cancers. The initial infectious process, i.e., the binding of the virus particle and its entry into the host cell, has been studied extensively, although it is not fully understood. There is still a gap in understanding the steps by which the virus is able to cross the plasma membrane after receptor binding. In this study, we demonstrate that after HPV16 comes into contact with a plasma membrane receptor, there are cytoskeletal changes resulting in an increase of filopodia numbers. This increase in filopodia numbers was transient and was maintained during the first two hours after virus addition. Our data show that there is a statistically significant increase in infection when filopodia numbers are increased by the addition of drug and virus simultaneously, and a decrease in virus infection when filopodia formation is inhibited. We describe that HPV16 binding results in the activation of Cdc42 GTPase that in turn results in an increase in filopodia. siRNA directed at Cdc42 GTPase resulted in a statistically significant reduction of infection and a corresponding lack of filopodia induction.
Keywords: HPV16; filopodia; viral entry.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
Similar articles
-
Vimentin Modulates Infectious Internalization of Human Papillomavirus 16 Pseudovirions.J Virol. 2017 Jul 27;91(16):e00307-17. doi: 10.1128/JVI.00307-17. Print 2017 Aug 15. J Virol. 2017. PMID: 28566373 Free PMC article.
-
Cross-talk between Rac1 and Cdc42 GTPases regulates formation of filopodia required for dengue virus type-2 entry into HMEC-1 cells.J Gen Virol. 2009 Dec;90(Pt 12):2902-2911. doi: 10.1099/vir.0.014159-0. Epub 2009 Aug 26. J Gen Virol. 2009. PMID: 19710257
-
Exogenous Vimentin Supplementation Transiently Affects Early Steps during HPV16 Pseudovirus Infection.Viruses. 2021 Dec 10;13(12):2471. doi: 10.3390/v13122471. Viruses. 2021. PMID: 34960740 Free PMC article.
-
The evolving field of human papillomavirus receptor research: a review of binding and entry.J Virol. 2013 Jun;87(11):6062-72. doi: 10.1128/JVI.00330-13. Epub 2013 Mar 27. J Virol. 2013. PMID: 23536685 Free PMC article. Review.
-
HPV16 Entry into Epithelial Cells: Running a Gauntlet.Viruses. 2021 Dec 8;13(12):2460. doi: 10.3390/v13122460. Viruses. 2021. PMID: 34960729 Free PMC article. Review.
Cited by
-
Human Papillomavirus Type 16 Stimulates WAVE1- and WAVE2-Dependent Actin Protrusions for Endocytic Entry.Viruses. 2025 Apr 8;17(4):542. doi: 10.3390/v17040542. Viruses. 2025. PMID: 40284985 Free PMC article.
References
-
- Chaturvedi A.K., Engels E.A., Pfeiffer R.M., Hernandez B.Y., Xiao W., Kim E., Jiang B., Goodman M.T., Sibug-Saber M., Cozen W., et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J. Clin. Oncol. 2011;29:4294–4301. doi: 10.1200/JCO.2011.36.4596. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
