

Classical Force-Field Parameters for CsPbBr3 Perovskite Nanocrystals
- PMID: 35747512
- PMCID: PMC9207923
- DOI: 10.1021/acs.jpcc.2c00600
Classical Force-Field Parameters for CsPbBr3 Perovskite Nanocrystals
Abstract
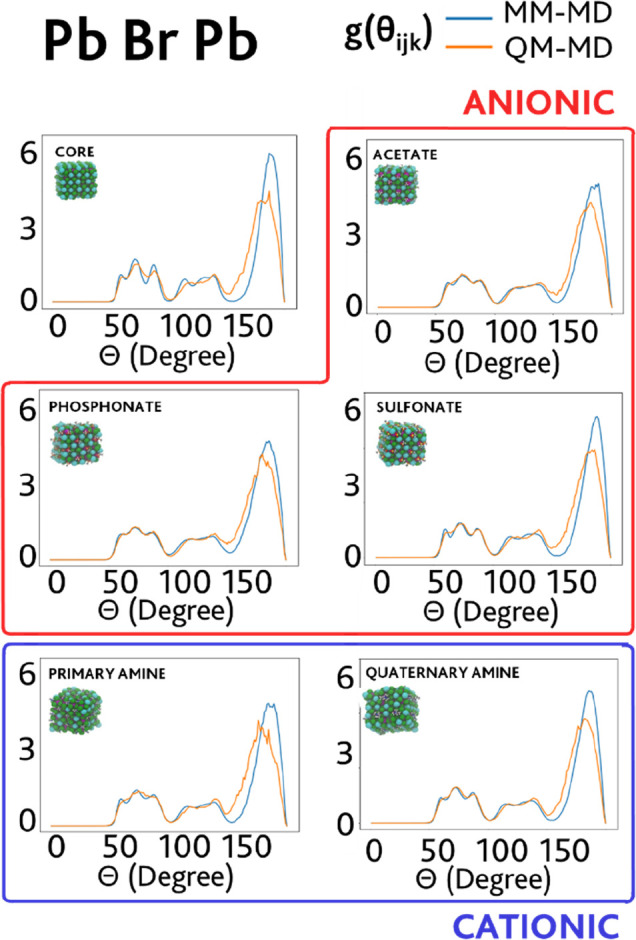
Understanding the chemico-physical properties of colloidal semiconductor nanocrystals (NCs) requires exploration of the dynamic processes occurring at the NC surfaces, in particular at the ligand-NC interface. Classical molecular dynamics (MD) simulations under realistic conditions are a powerful tool to acquire this knowledge because they have good accuracy and are computationally cheap, provided that a set of force-field (FF) parameters is available. In this work, we employed a stochastic algorithm, the adaptive rate Monte Carlo method, to optimize FF parameters of cesium lead halide perovskite (CsPbBr3) NCs passivated with typical organic molecules used in the synthesis of these materials: oleates, phosphonates, sulfonates, and primary and quaternary ammonium ligands. The optimized FF parameters have been obtained against MD reference trajectories computed at the density functional theory level on small NC model systems. We validated our parameters through a comparison of a wide range of nonfitted properties to experimentally available values. With the exception of the NC-phosphonate case, the transferability of the FF model has been successfully tested on realistically sized systems (>5 nm) comprising thousands of passivating organic ligands and solvent molecules, just as those used in experiments.
© 2022 The Authors. Published by American Chemical Society.
Conflict of interest statement
The authors declare no competing financial interest.
Figures





References
-
- Hetsch F.; Xu X.; Wang H.; Kershaw S. V.; Rogach A. L. Semiconductor Nanocrystal Quantum Dots as Solar Cell Components and Photosensitizers: Material, Charge Transfer, and Separation Aspects of Some Device Topologies. J. Phys. Chem. Lett. 2011, 2, 1879. 10.1021/jz200802j. - DOI
-
- Sargent E. H.Colloidal Quantum Dot Solar Cells; 2012; Vol. 6. 10.1038/nphoton.2012.33. - DOI
LinkOut - more resources
Full Text Sources