

Detection of SARS-CoV-2 intra-host recombination during superinfection with Alpha and Epsilon variants in New York City
- PMID: 35752633
- PMCID: PMC9233664
- DOI: 10.1038/s41467-022-31247-x
Detection of SARS-CoV-2 intra-host recombination during superinfection with Alpha and Epsilon variants in New York City
Abstract
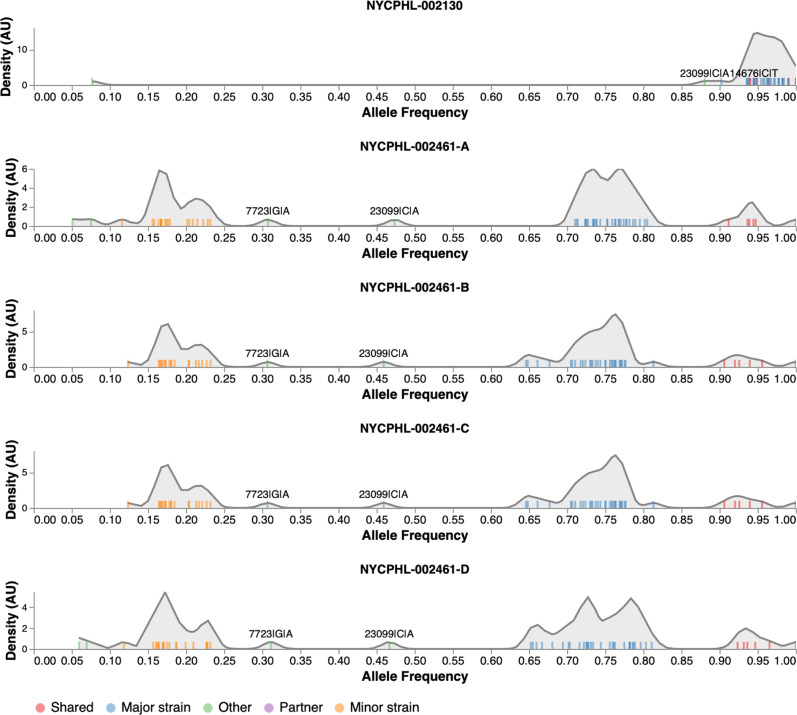
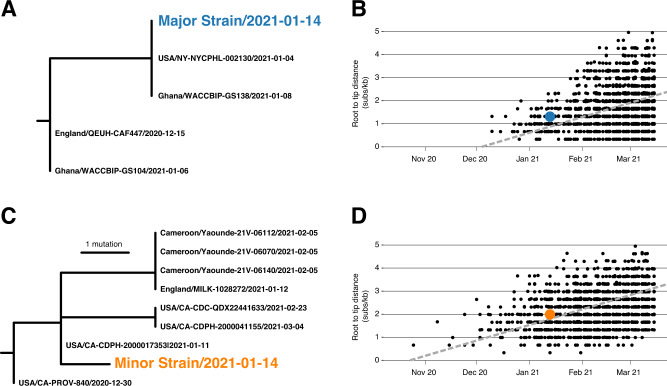
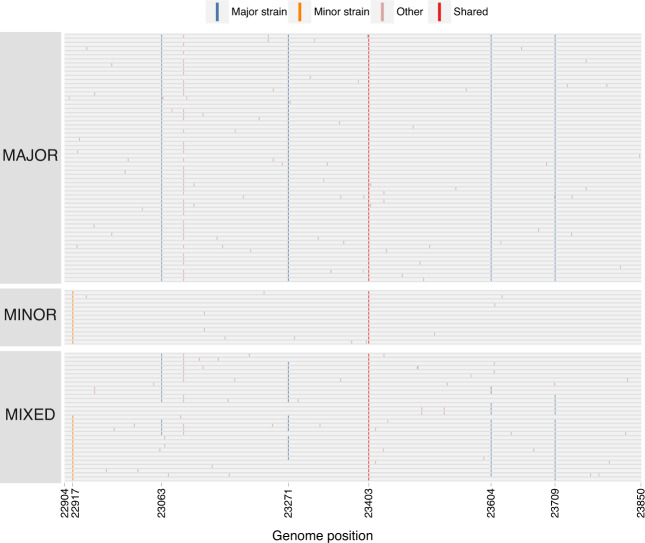
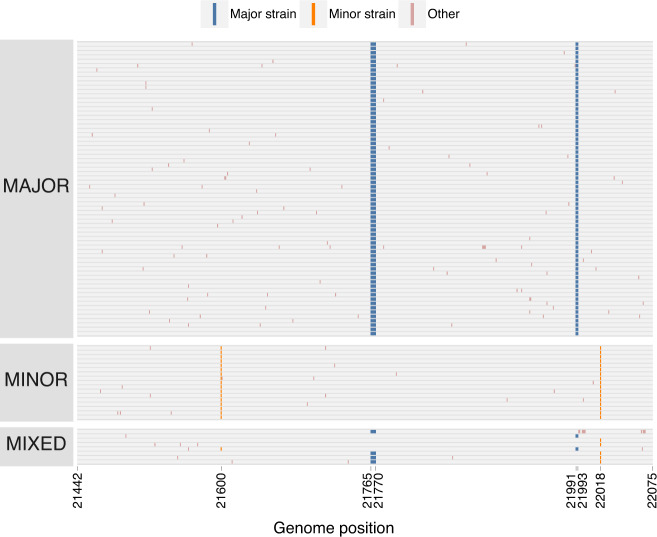
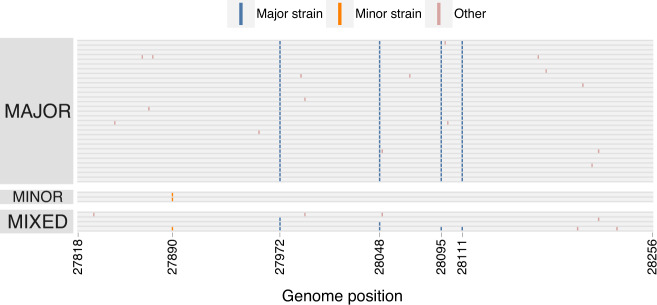
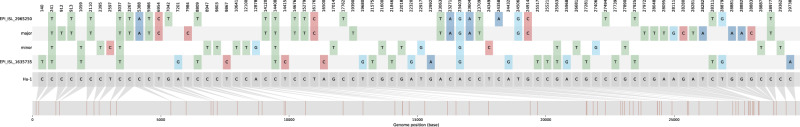
Recombination is an evolutionary process by which many pathogens generate diversity and acquire novel functions. Although a common occurrence during coronavirus replication, detection of recombination is only feasible when genetically distinct viruses contemporaneously infect the same host. Here, we identify an instance of SARS-CoV-2 superinfection, whereby an individual was infected with two distinct viral variants: Alpha (B.1.1.7) and Epsilon (B.1.429). This superinfection was first noted when an Alpha genome sequence failed to exhibit the classic S gene target failure behavior used to track this variant. Full genome sequencing from four independent extracts reveals that Alpha variant alleles comprise around 75% of the genomes, whereas the Epsilon variant alleles comprise around 20% of the sample. Further investigation reveals the presence of numerous recombinant haplotypes spanning the genome, specifically in the spike, nucleocapsid, and ORF 8 coding regions. These findings support the potential for recombination to reshape SARS-CoV-2 genetic diversity.
© 2022. The Author(s).
Conflict of interest statement
J.O.W. and S.L.K.P. has received funding from the CDC (ongoing) via contracts or agreements to their institution unrelated to this research. The remaining authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
