

Sophoridine Suppresses Herpes Simplex Virus Type 1 Infection by Blocking the Activation of Cellular PI3K/Akt and p38 MAPK Pathways
- PMID: 35756044
- PMCID: PMC9229184
- DOI: 10.3389/fmicb.2022.872505
Sophoridine Suppresses Herpes Simplex Virus Type 1 Infection by Blocking the Activation of Cellular PI3K/Akt and p38 MAPK Pathways
Abstract
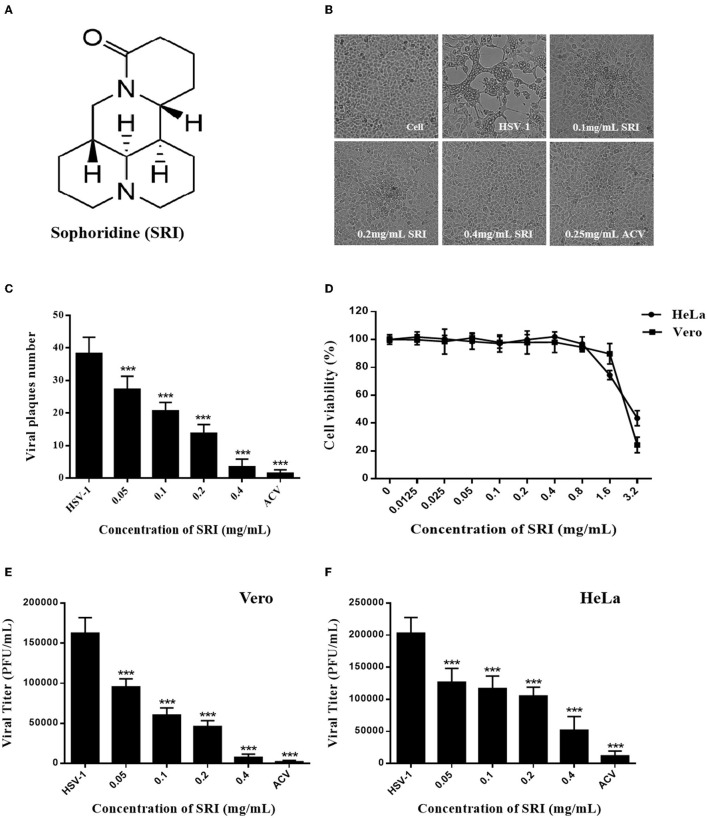
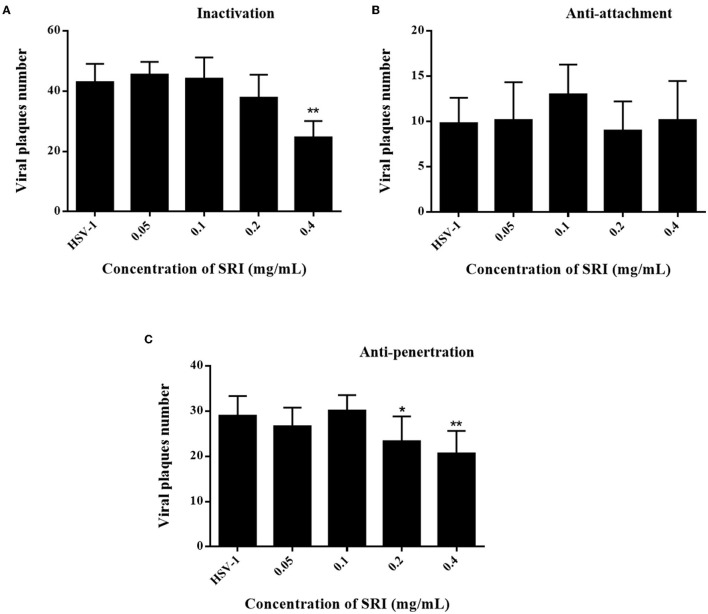
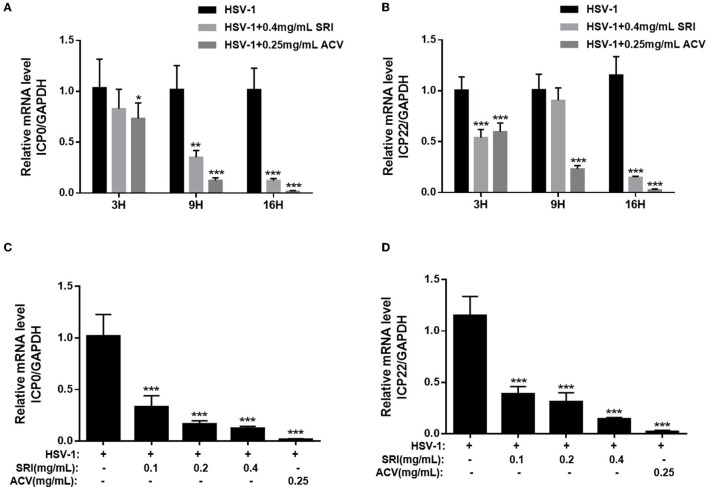
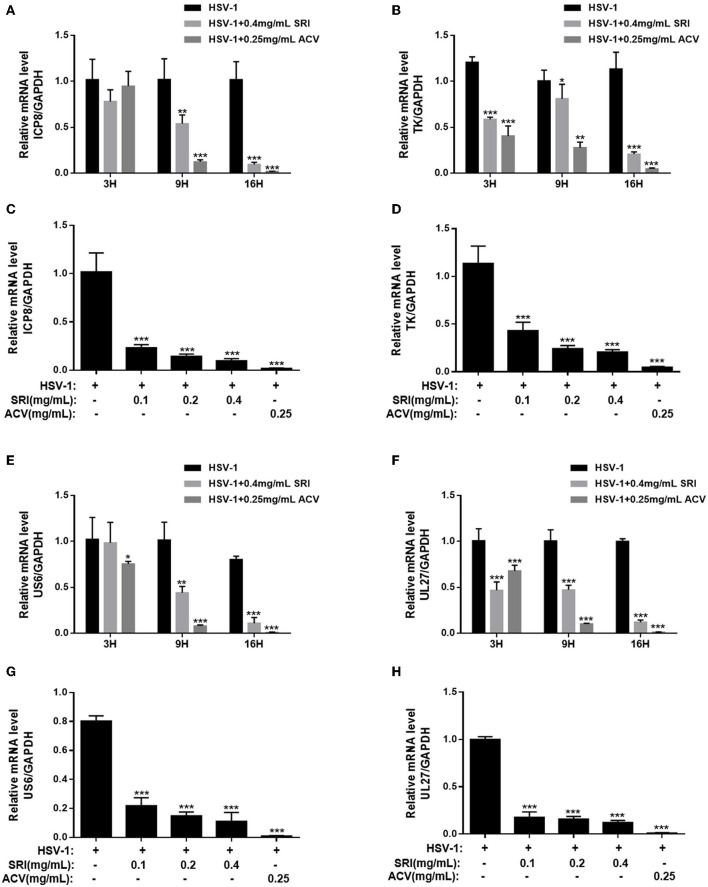
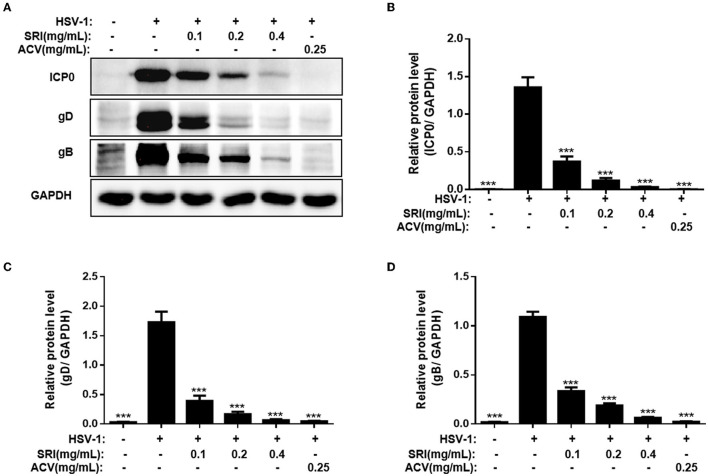
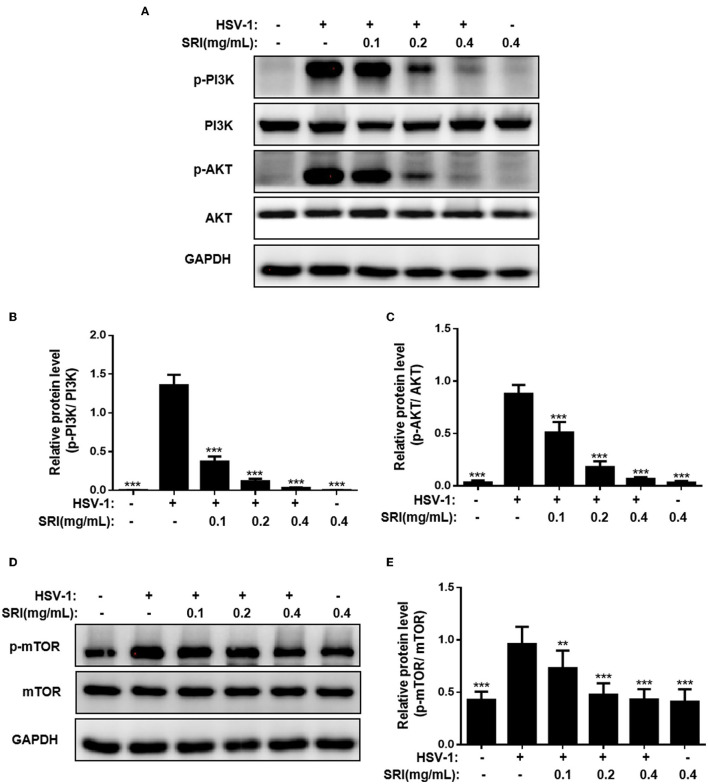
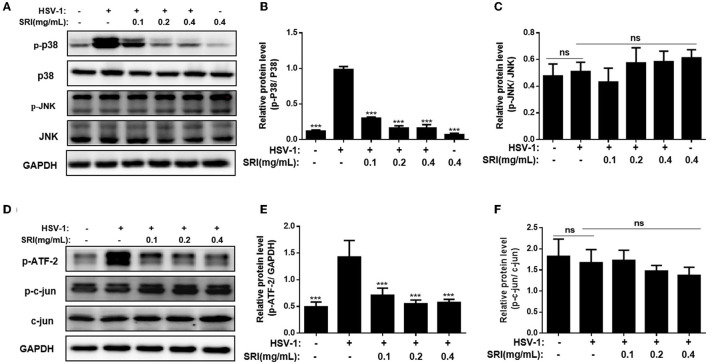
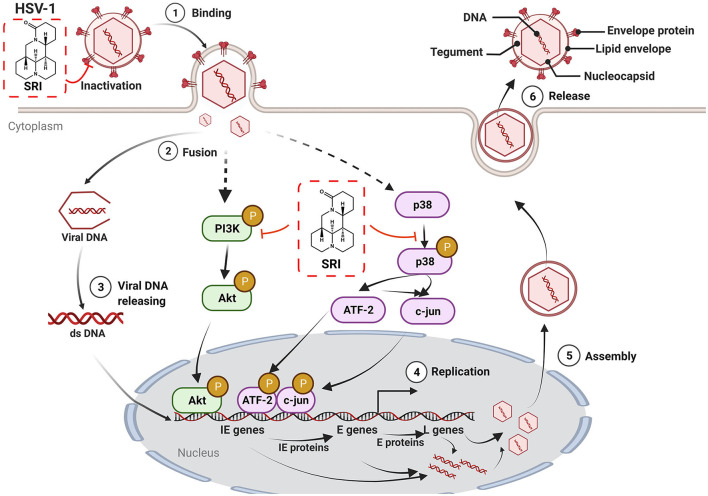
Herpes simplex virus type 1 (HSV-1) is a ubiquitous and important human pathogen capable of causing significant clinical diseases ranging from skin damage to encephalitis, particularly in immunocompromised and neonatal hosts. Currently, widely used nucleoside analogs, including acyclovir and penciclovir, have some limitations in their use due to side effects and drug resistance. Herein, we report sophoridine's (SRI) dramatic inhibition of HSV-1 replication in vitro. SRI exhibited a remarkable inhibitory influence on HSV-1 virus-induced cytopathic effect and plaque formation, as well as on progeny viruses in Vero and HeLa cells, with selection indexes (SI) of 38.96 and 22.62, respectively. Moreover, SRI also considerably suppressed HSV-1 replication by hindering the expression of viral immediate-early (ICP0 and ICP22), early (ICP8 and TK), and late (gB and gD) genes and the expression of viral proteins ICP0, gB, and gD. We suggest that SRI can directly inactivate viral particles and block some stages in the life cycle of HSV-1 after adsorption. Further experiments showed that SRI downregulated the cellular PI3K/Akt signaling pathway and obstructed HSV-1 replication even more. Most importantly, SRI markedly repressed HSV-1-induced p38 MAPK pathway activation. Collectively, this natural bioactive alkaloid could be a promising therapeutic candidate against HSV-1 via the modulation of cellular PI3K/Akt and p38 MAPK pathways.
Keywords: HSV-1; PI3K/Akt pathway; antiviral; p38 MAPK pathway; sophoridine.
Copyright © 2022 Tang, Luan, Yuan, Sun, Rao, Wang, Liu and Zeng.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
LinkOut - more resources
Full Text Sources