

Markov chains improve the significance computation of overlapping genome annotations
- PMID: 35758770
- PMCID: PMC9235476
- DOI: 10.1093/bioinformatics/btac255
Markov chains improve the significance computation of overlapping genome annotations
Abstract
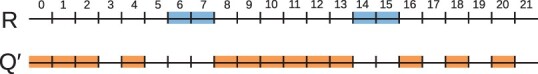
Motivation: Genome annotations are a common way to represent genomic features such as genes, regulatory elements or epigenetic modifications. The amount of overlap between two annotations is often used to ascertain if there is an underlying biological connection between them. In order to distinguish between true biological association and overlap by pure chance, a robust measure of significance is required. One common way to do this is to determine if the number of intervals in the reference annotation that intersect the query annotation is statistically significant. However, currently employed statistical frameworks are often either inefficient or inaccurate when computing P-values on the scale of the whole human genome.
Results: We show that finding the P-values under the typically used 'gold' null hypothesis is NP-hard. This motivates us to reformulate the null hypothesis using Markov chains. To be able to measure the fidelity of our Markovian null hypothesis, we develop a fast direct sampling algorithm to estimate the P-value under the gold null hypothesis. We then present an open-source software tool MCDP that computes the P-values under the Markovian null hypothesis in O(m2+n) time and O(m) memory, where m and n are the numbers of intervals in the reference and query annotations, respectively. Notably, MCDP runtime and memory usage are independent from the genome length, allowing it to outperform previous approaches in runtime and memory usage by orders of magnitude on human genome annotations, while maintaining the same level of accuracy.
Availability and implementation: The software is available at https://github.com/fmfi-compbio/mc-overlaps. All data for reproducibility are available at https://github.com/fmfi-compbio/mc-overlaps-reproducibility.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2022. Published by Oxford University Press.
Figures



References
-
- Burns J. (1983). If nothing goes wrong, is everything all right? Why we should be wary of zero numerators. J. Am. Med. Assoc., 249(13), 1743–1745. - PubMed
-
- Devroye L. (1986). Non-Uniform Random Variate Generation. Springer, New York.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
