

TRPM7 promotes lipopolysaccharide-induced inflammatory dysfunction in renal tubular epithelial cells
- PMID: 35759233
- PMCID: PMC9208284
- DOI: 10.1002/iid3.641
TRPM7 promotes lipopolysaccharide-induced inflammatory dysfunction in renal tubular epithelial cells
Abstract
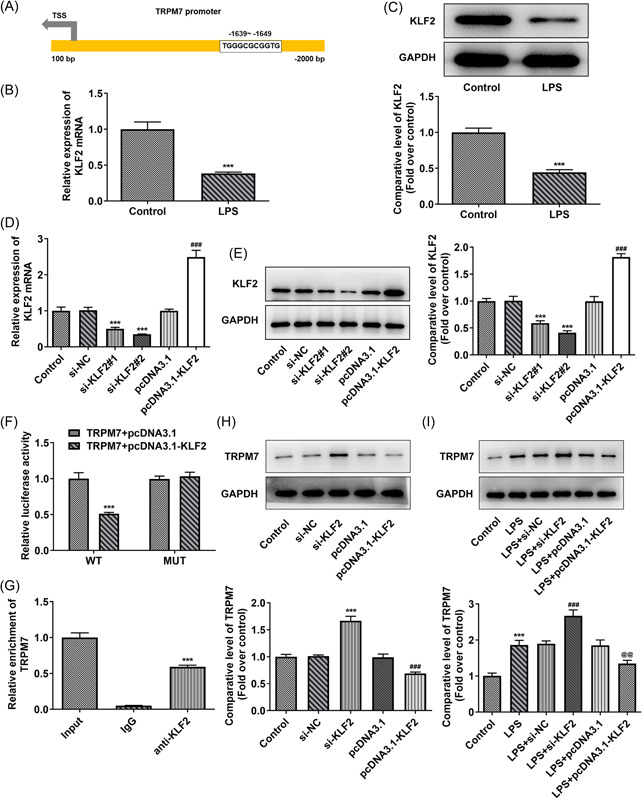
Background: Sepsis-associated acute kidney injury (S-AKI) has been reported to affect 30%-50% of all sepsis patients; this condition is associated with a notable fatality rate. Following lipopolysaccharide (LPS) stimulation, the expression of transient receptor potential cation channel subfamily M member 7 (TRPM7), a nonselective cation channel expressed by the renal tubular epithelial cells (RTECs) was found to be upregulated. We aimed to determine how TRPM7 functions in S-AKI.
Methods: To establish an in vitro model of S-AKI, RTECs were treated with LPS. The effect of TRPM7 knockdown on cell viability, lactate dehydrogenase (LDH) release, apoptosis, inflammation, and oxidative stress was studied. The binding site between Kruppel-like factor 2 (KLF2) and TRPM7 was predicted using JASPAR. The influence of KLF2 on the regulatory roles of TRPM7 in cells, as well as the effect of their knockdown on the MAPK signaling pathway, was investigated.
Results: TRPM7 was upregulated in LPS-treated cells, and knocking improved cell viability, reduced LDH levels, and minimized apoptosis, inflammation, and oxidative stress. KLF2 was shown to be associated with TRPM7 and its level decreased in LPS-treated cells. KLF2 knockdown increased TRPM7 expression and reversed the effects of TRPM7 knockdown in LPS-treated cells, including suppression of p38 MAPK, ERK1/2, and JNK activation.
Conclusion: Taken together, our results show that TRPM7 is negatively regulated by KLF2 and promotes LPS-induced inflammatory dysfunction by activating the MAPK pathway in RTECs. The theoretical foundation for the prevention and management of S-AKI is laid out in this article.
Keywords: Kruppel-like factor 2; TRPM7; inflammation; renal tubular epithelial cells; sepsis.
© 2022 John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures





References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
