

Genome sequence diversity of SARS-CoV-2 obtained from clinical samples in Uzbekistan
- PMID: 35759503
- PMCID: PMC9236271
- DOI: 10.1371/journal.pone.0270314
Genome sequence diversity of SARS-CoV-2 obtained from clinical samples in Uzbekistan
Abstract
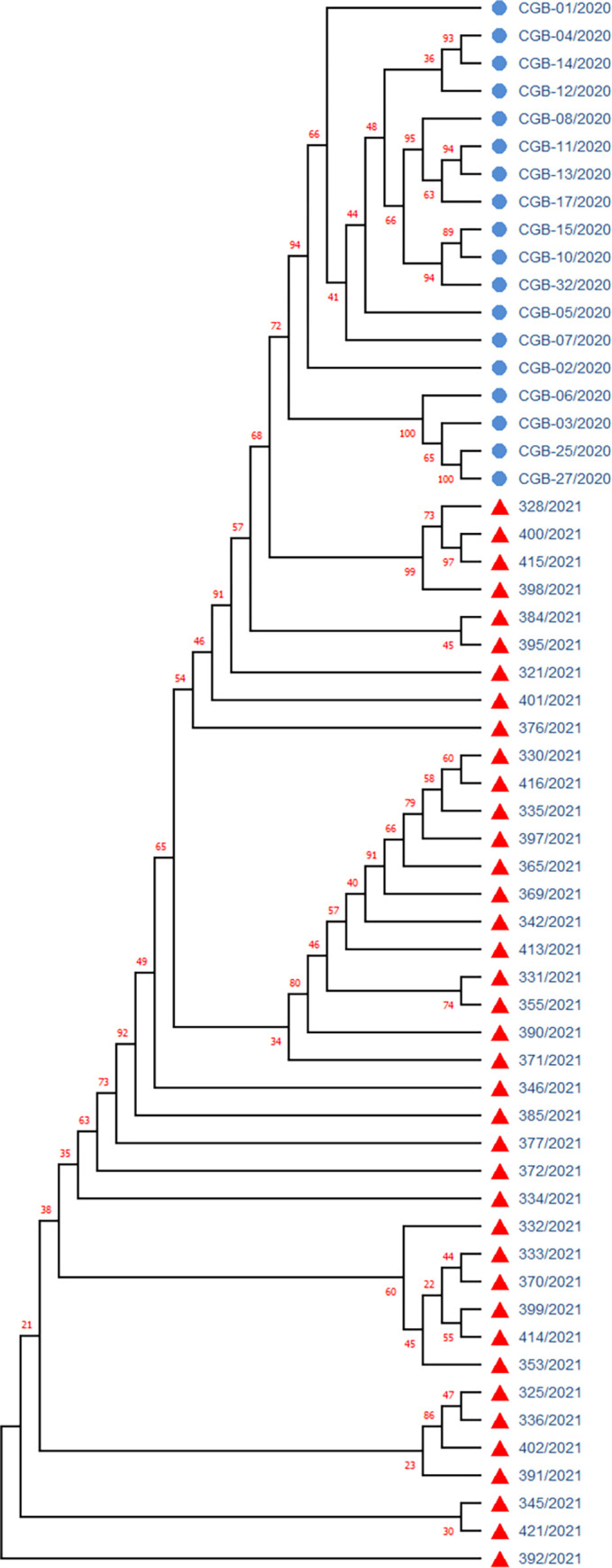
Tracking temporal and spatial genomic changes and evolution of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) are among the most urgent research topics worldwide, which help to elucidate the coronavirus disease 2019 (COVID-19) pathogenesis and the effect of deleterious variants. Our current study concentrates genetic diversity of SARS-CoV-2 variants in Uzbekistan and their associations with COVID-19 severity. Thirty-nine whole genome sequences (WGS) of SARS-CoV-2 isolated from PCR-positive patients from Tashkent, Uzbekistan for the period of July-August 2021, were generated and further subjected to further genomic analysis. Genome-wide annotations of clinical isolates from our study have revealed a total of 223 nucleotide-level variations including SNPs and 34 deletions at different positions throughout the entire genome of SARS-CoV-2. These changes included two novel mutations at the Nonstructural protein (Nsp) 13: A85P and Nsp12: Y479N, which were unreported previously. There were two groups of co-occurred substitution patterns: the missense mutations in the Spike (S): D614G, Open Reading Frame (ORF) 1b: P314L, Nsp3: F924, 5`UTR:C241T; Nsp3:P2046L and Nsp3:P2287S, and the synonymous mutations in the Nsp4:D2907 (C8986T), Nsp6:T3646A and Nsp14:A1918V regions, respectively. The "Nextstrain" clustered the largest number of SARS-CoV-2 strains into the Delta clade (n = 32; 82%), followed by two Alpha-originated (n = 4; 10,3%) and 20A (n = 3; 7,7%) clades. Geographically the Delta clade sample sequences were grouped into several clusters with the SARS-CoV genotypes from Russia, Denmark, USA, Egypt and Bangladesh. Phylogenetically, the Delta isolates in our study belong to the two main subclades 21A (56%) and 21J (44%). We found that females were more affected by 21A, whereas males by 21J variant (χ2 = 4.57; p ≤ 0.05, n = 32). The amino acid substitution ORF7a:P45L in the Delta isolates found to be significantly associated with disease severity. In conclusion, this study evidenced that Identified novel substitutions Nsp13: A85P and Nsp12: Y479N, have a destabilizing effect, while missense substitution ORF7a: P45L significantly associated with disease severity.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures











References
-
- COVID-19 Map—Johns Hopkins Coronavirus Resource Center. [cited 16 Sep 2021]. Available: https://coronavirus.jhu.edu/map.html
Publication types
MeSH terms
Substances
Supplementary concepts
Associated data
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
