

Activating STING1-dependent immune signaling in TP53 mutant and wild-type acute myeloid leukemia
- PMID: 35759659
- PMCID: PMC9271208
- DOI: 10.1073/pnas.2123227119
Activating STING1-dependent immune signaling in TP53 mutant and wild-type acute myeloid leukemia
Abstract
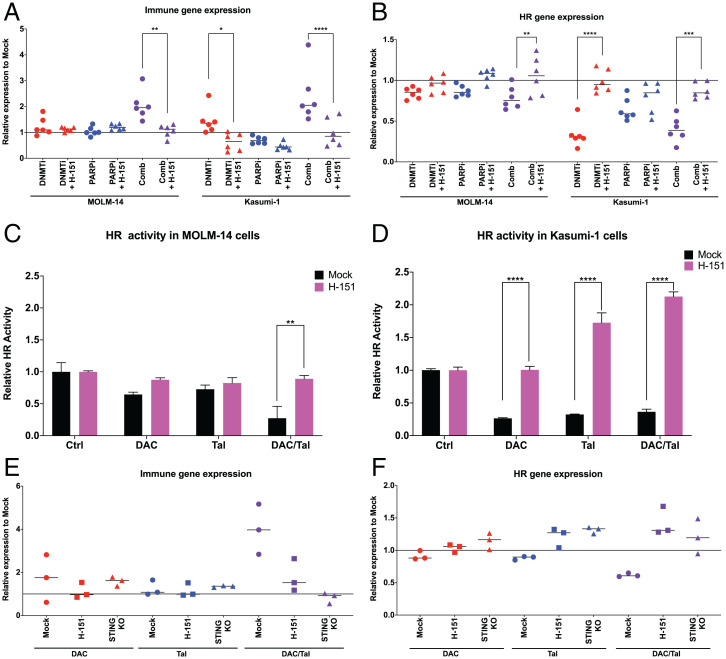
DNA methyltransferase inhibitors (DNMTis) reexpress hypermethylated genes in cancers and leukemias and also activate endogenous retroviruses (ERVs), leading to interferon (IFN) signaling, in a process known as viral mimicry. In the present study we show that in the subset of acute myeloid leukemias (AMLs) with mutations in TP53, associated with poor prognosis, DNMTis, important drugs for treatment of AML, enable expression of ERVs and IFN and inflammasome signaling in a STING-dependent manner. We previously reported that in solid tumors poly ADP ribose polymerase inhibitors (PARPis) combined with DNMTis to induce an IFN/inflammasome response that is dependent on STING1 and is mechanistically linked to generation of a homologous recombination defect (HRD). We now show that STING1 activity is actually increased in TP53 mutant compared with wild-type (WT) TP53 AML. Moreover, in TP53 mutant AML, STING1-dependent IFN/inflammatory signaling is increased by DNMTi treatment, whereas in AMLs with WT TP53, DNMTis alone have no effect. While combining DNMTis with PARPis increases IFN/inflammatory gene expression in WT TP53 AML cells, signaling induced in TP53 mutant AML is still several-fold higher. Notably, induction of HRD in both TP53 mutant and WT AMLs follows the pattern of STING1-dependent IFN and inflammatory signaling that we have observed with drug treatments. These findings increase our understanding of the mechanisms that underlie DNMTi + PARPi treatment, and also DNMTi combinations with immune therapies, suggesting a personalized approach that statifies by TP53 status, for use of such therapies, including potential immune activation of STING1 in AML and other cancers.
Keywords: AML; TP53; combination therapy; epigenetics; immune signaling.
Conflict of interest statement
The authors declare a competing interest. F.V.R. and S.B.B. share co-inventorship on US Provisional Patent Application No. 61/929,680 for the concept of the combinatorial therapy.
Figures






References
-
- Kogan A. A., Lapidus R. G., Baer M. R., Rassool F. V., Exploiting epigenetically mediated changes: Acute myeloid leukemia, leukemia stem cells and the bone marrow microenvironment. Adv. Cancer Res. 141, 213–253 (2019). - PubMed
-
- Bullinger L., Döhner K., Döhner H., Genomics of acute myeloid leukemia diagnosis and pathways. J. Clin. Oncol. 35, 934–946 (2017). - PubMed
-
- Bowen D., et al. , TP53 gene mutation is frequent in patients with acute myeloid leukemia and complex karyotype, and is associated with very poor prognosis. Leukemia 23, 203–206 (2009). - PubMed
-
- Haferlach C., et al. , Mutations of the TP53 gene in acute myeloid leukemia are strongly associated with a complex aberrant karyotype. Leukemia 22, 1539–1541 (2008). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
