

An introduction to spatial transcriptomics for biomedical research
- PMID: 35761361
- PMCID: PMC9238181
- DOI: 10.1186/s13073-022-01075-1
An introduction to spatial transcriptomics for biomedical research
Abstract
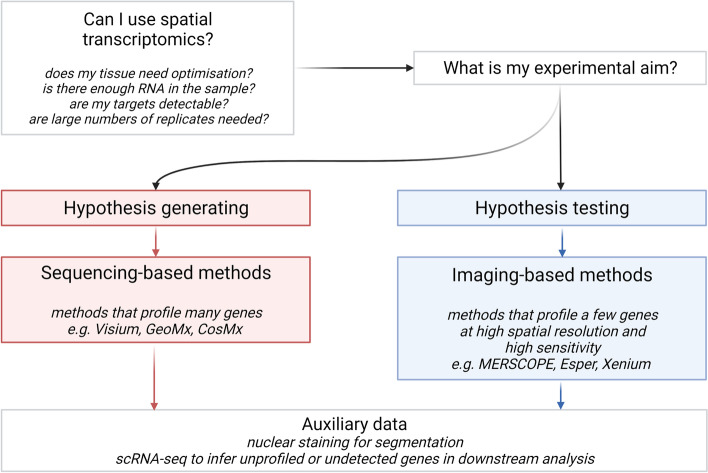
Single-cell transcriptomics (scRNA-seq) has become essential for biomedical research over the past decade, particularly in developmental biology, cancer, immunology, and neuroscience. Most commercially available scRNA-seq protocols require cells to be recovered intact and viable from tissue. This has precluded many cell types from study and largely destroys the spatial context that could otherwise inform analyses of cell identity and function. An increasing number of commercially available platforms now facilitate spatially resolved, high-dimensional assessment of gene transcription, known as 'spatial transcriptomics'. Here, we introduce different classes of method, which either record the locations of hybridized mRNA molecules in tissue, image the positions of cells themselves prior to assessment, or employ spatial arrays of mRNA probes of pre-determined location. We review sizes of tissue area that can be assessed, their spatial resolution, and the number and types of genes that can be profiled. We discuss if tissue preservation influences choice of platform, and provide guidance on whether specific platforms may be better suited to discovery screens or hypothesis testing. Finally, we introduce bioinformatic methods for analysing spatial transcriptomic data, including pre-processing, integration with existing scRNA-seq data, and inference of cell-cell interactions. Spatial -omics methods are already improving our understanding of human tissues in research, diagnostic, and therapeutic settings. To build upon these recent advancements, we provide entry-level guidance for those seeking to employ spatial transcriptomics in their own biomedical research.
© 2022. The Author(s).
Conflict of interest statement
CGW owns shares in 10X Genomics. The remaining authors declare that they have no competing interests.
Figures


References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
