

A critical evaluation of Mycobacterium bovis pangenomics, with reference to its utility in outbreak investigation
- PMID: 35763423
- PMCID: PMC9455707
- DOI: 10.1099/mgen.0.000839
A critical evaluation of Mycobacterium bovis pangenomics, with reference to its utility in outbreak investigation
Abstract
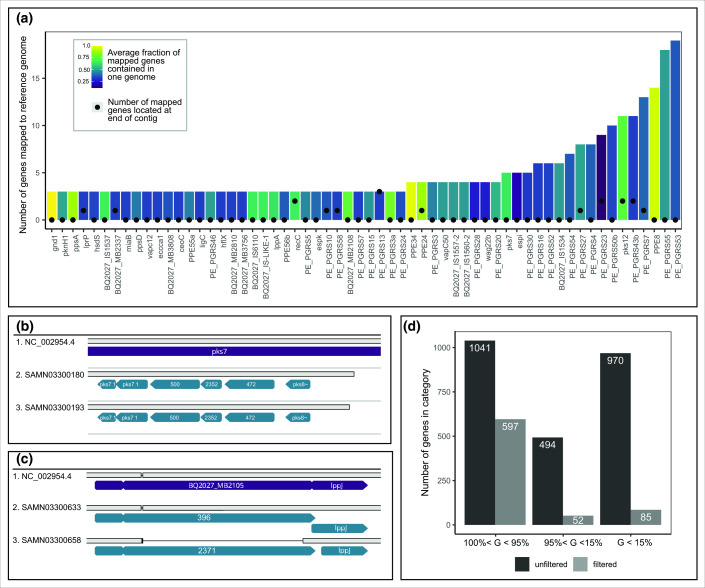
The increased accessibility of next generation sequencing has allowed enough genomes from a given bacterial species to be sequenced to describe the distribution of genes in the pangenome, without limiting analyses to genes present in reference strains. Although some taxa have thousands of whole genome sequences available on public databases, most genomes were sequenced with short read technology, resulting in incomplete assemblies. Studying pangenomes could lead to important insights into adaptation, pathogenicity, or molecular epidemiology, however given the known information loss inherent in analyzing contig-level assemblies, these inferences may be biased or inaccurate. In this study we describe the pangenome of a clonally evolving pathogen,
Keywords: Mycobacterium tuberculosis complex; molecular epidemiology; pangenomics.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures





References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources