

Whole-genome CRISPR screening identifies N-glycosylation as a genetic and therapeutic vulnerability in CALR-mutant MPNs
- PMID: 35763665
- PMCID: PMC9479036
- DOI: 10.1182/blood.2022015629
Whole-genome CRISPR screening identifies N-glycosylation as a genetic and therapeutic vulnerability in CALR-mutant MPNs
Erratum in
-
Jutzi JS, Marneth AE, Ciboddo M, et al. Whole-genome CRISPR screening identifies N-glycosylation as a genetic and therapeutic vulnerability in CALR-mutant MPNs. Blood. 2022;140(11):1291-1304.Blood. 2023 Nov 16;142(20):1758. doi: 10.1182/blood.2023022676. Blood. 2023. PMID: 37971758 Free PMC article. No abstract available.
Abstract
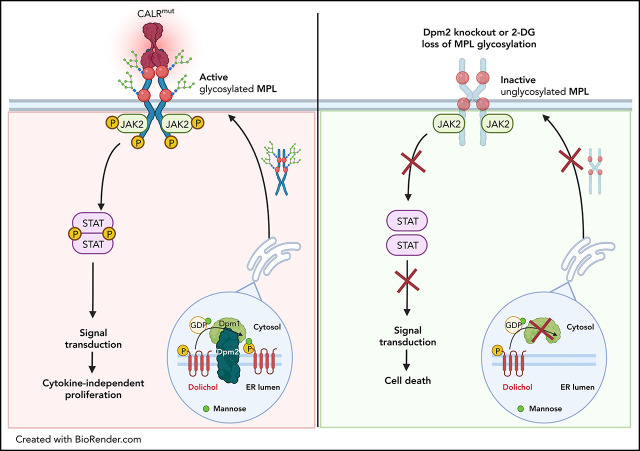
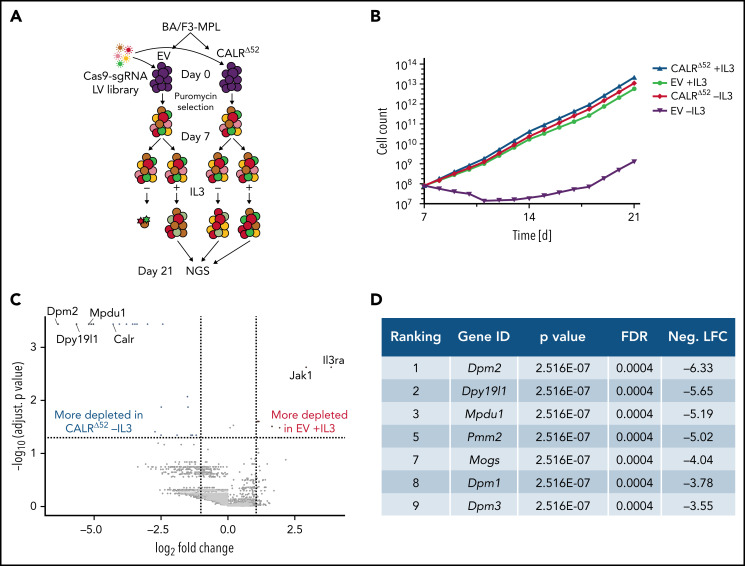
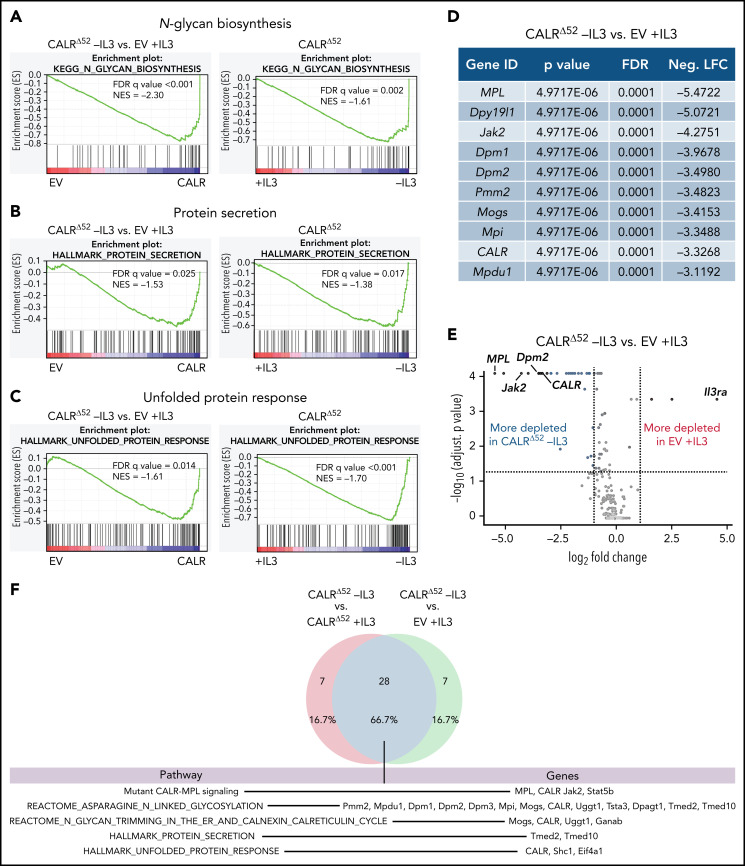
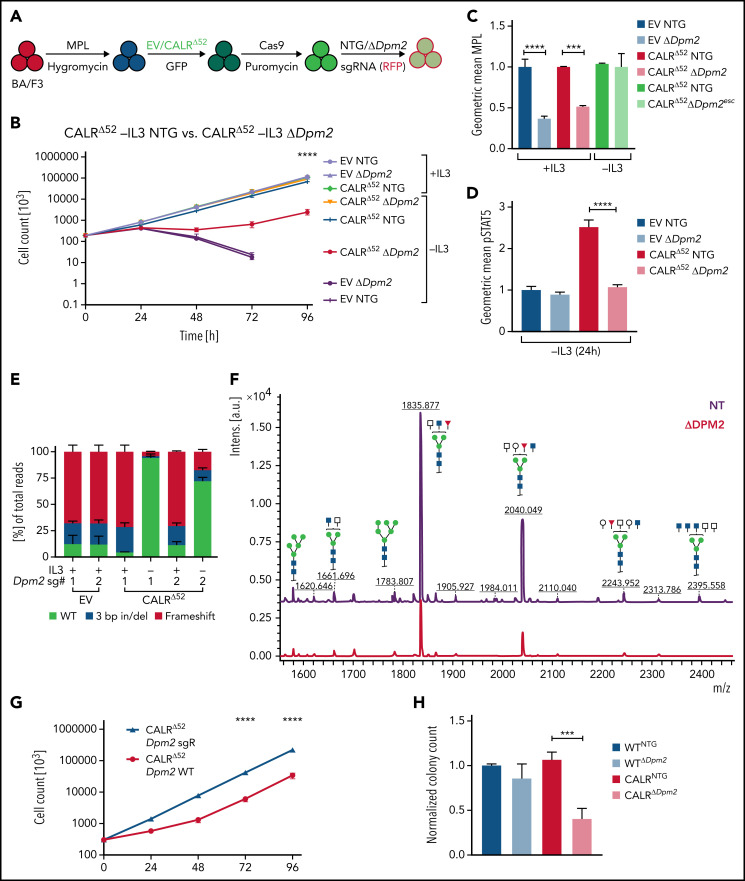
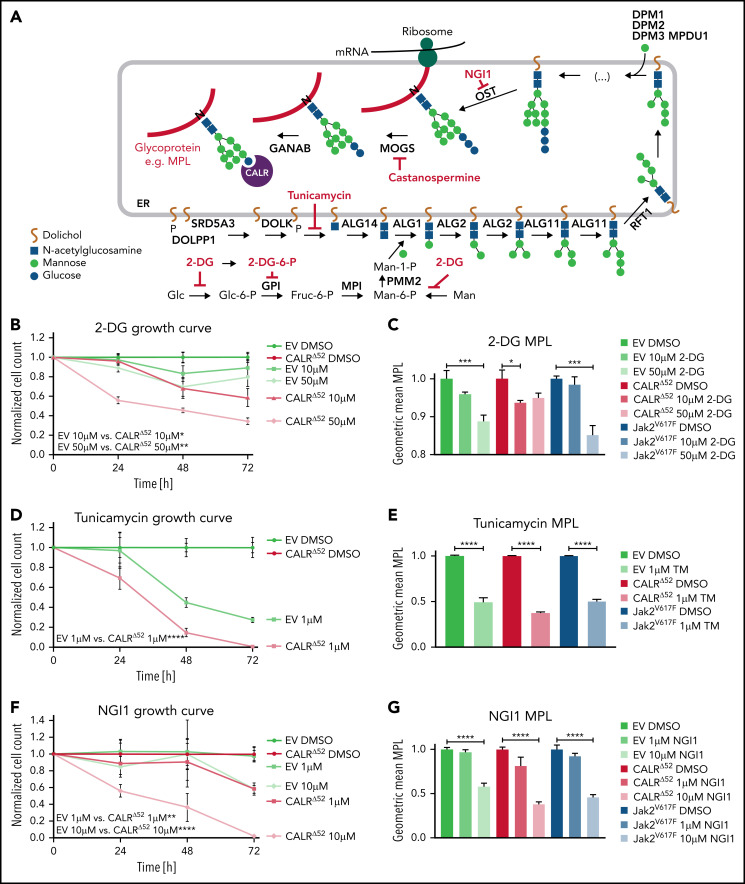
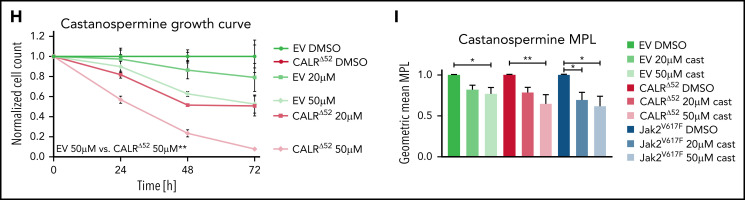
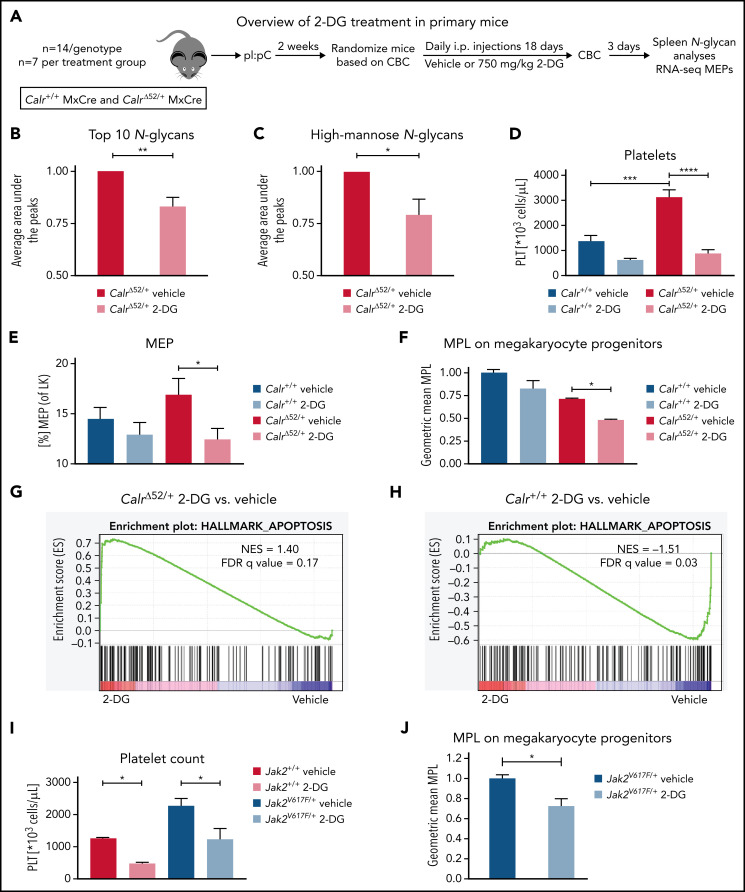
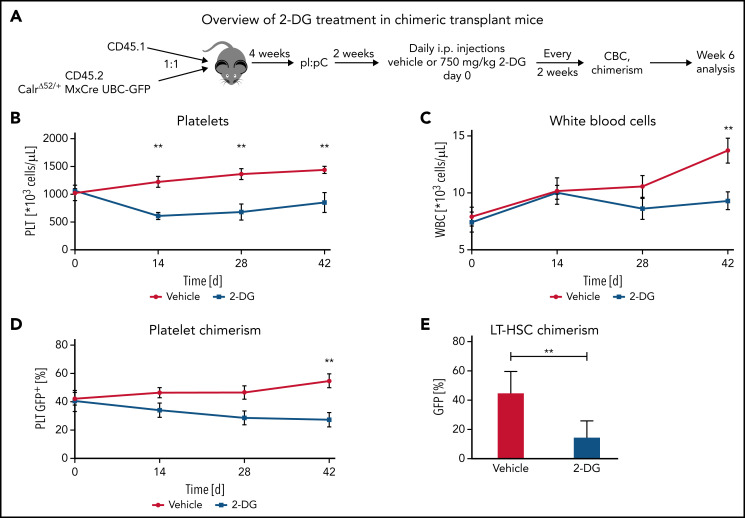
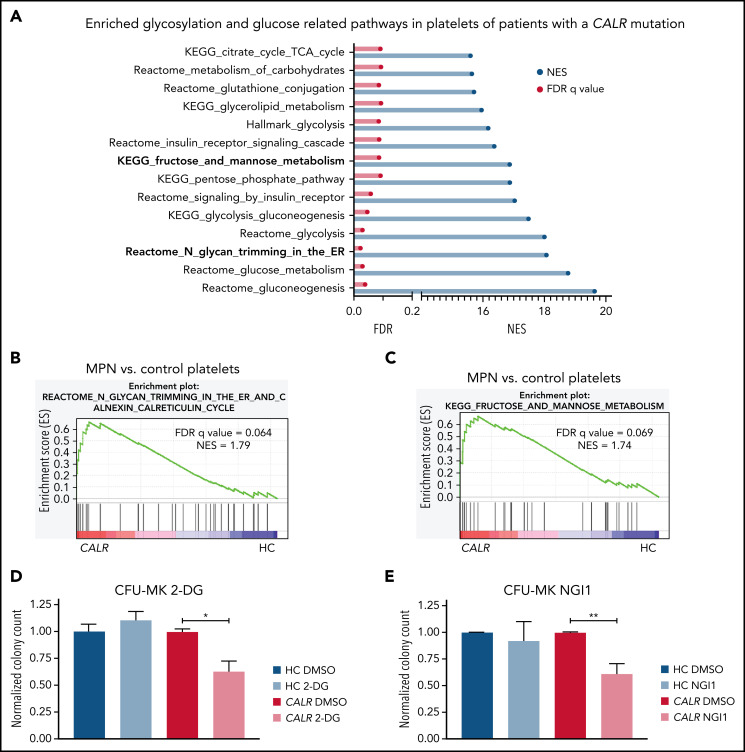
Calreticulin (CALR) mutations are frequent, disease-initiating events in myeloproliferative neoplasms (MPNs). Although the biological mechanism by which CALR mutations cause MPNs has been elucidated, there currently are no clonally selective therapies for CALR-mutant MPNs. To identify unique genetic dependencies in CALR-mutant MPNs, we performed a whole-genome clustered regularly interspaced short palindromic repeats (CRISPR) knockout depletion screen in mutant CALR-transformed hematopoietic cells. We found that genes in the N-glycosylation pathway (among others) were differentially depleted in mutant CALR-transformed cells as compared with control cells. Using a focused pharmacological in vitro screen targeting unique vulnerabilities uncovered in the CRISPR screen, we found that chemical inhibition of N-glycosylation impaired the growth of mutant CALR-transformed cells, through a reduction in MPL cell surface expression. We treated Calr-mutant knockin mice with the N-glycosylation inhibitor 2-deoxy-glucose (2-DG) and found a preferential sensitivity of Calr-mutant cells to 2-DG as compared with wild-type cells and normalization of key MPNs disease features. To validate our findings in primary human cells, we performed megakaryocyte colony-forming unit (CFU-MK) assays. We found that N-glycosylation inhibition significantly reduced CFU-MK formation in patient-derived CALR-mutant bone marrow as compared with bone marrow derived from healthy donors. In aggregate, our findings advance the development of clonally selective treatments for CALR-mutant MPNs.
© 2022 by The American Society of Hematology.
Figures
Comment in
-
Mutant CALR's "sweet tooth".Blood. 2022 Sep 15;140(11):1187-1189. doi: 10.1182/blood.2022017448. Blood. 2022. PMID: 36107460 Free PMC article. No abstract available.
References
-
- Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379-2390. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
