

Tumor-targeted interleukin-12 synergizes with entinostat to overcome PD-1/PD-L1 blockade-resistant tumors harboring MHC-I and APM deficiencies
- PMID: 35764364
- PMCID: PMC9240938
- DOI: 10.1136/jitc-2022-004561
Tumor-targeted interleukin-12 synergizes with entinostat to overcome PD-1/PD-L1 blockade-resistant tumors harboring MHC-I and APM deficiencies
Abstract
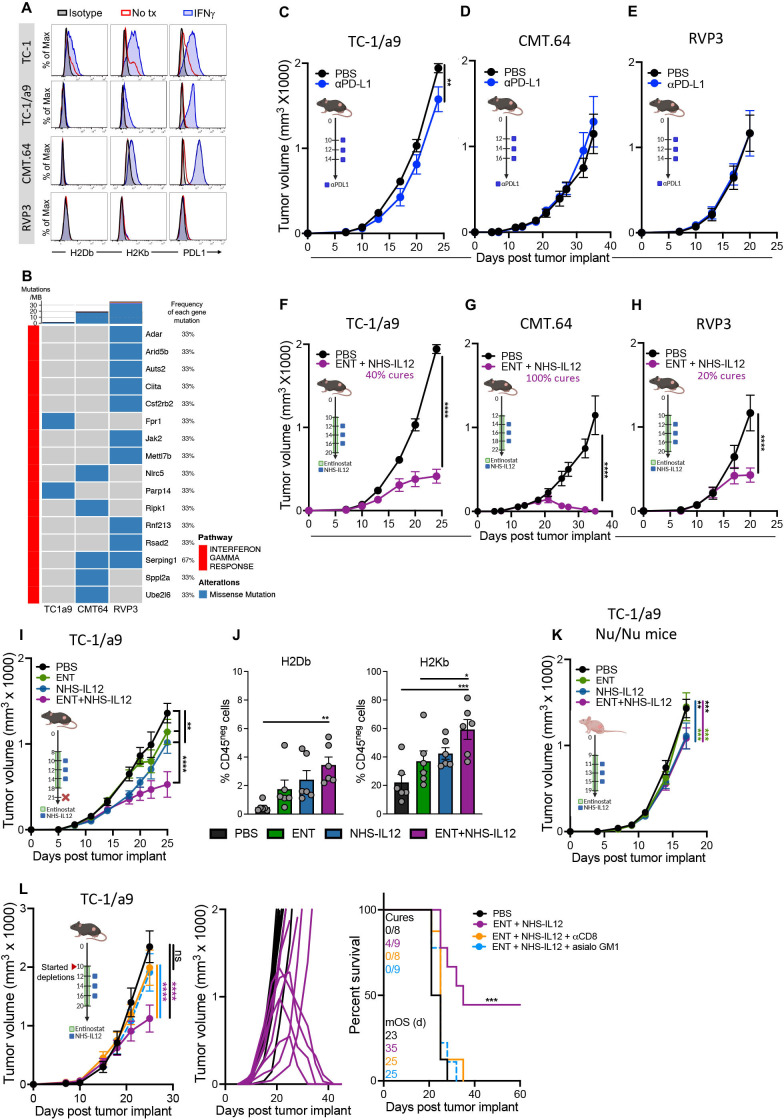
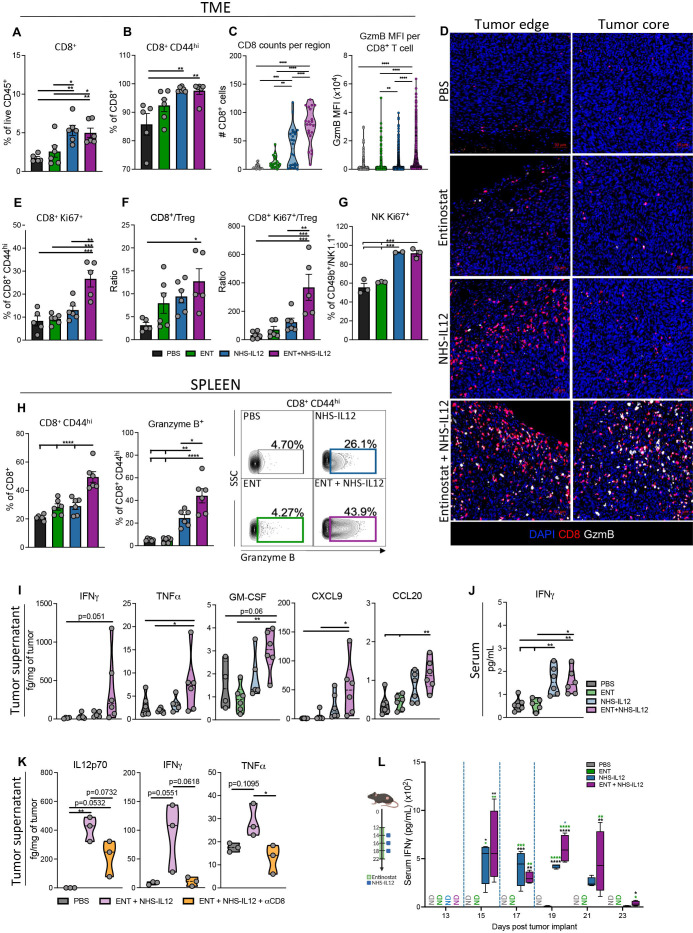
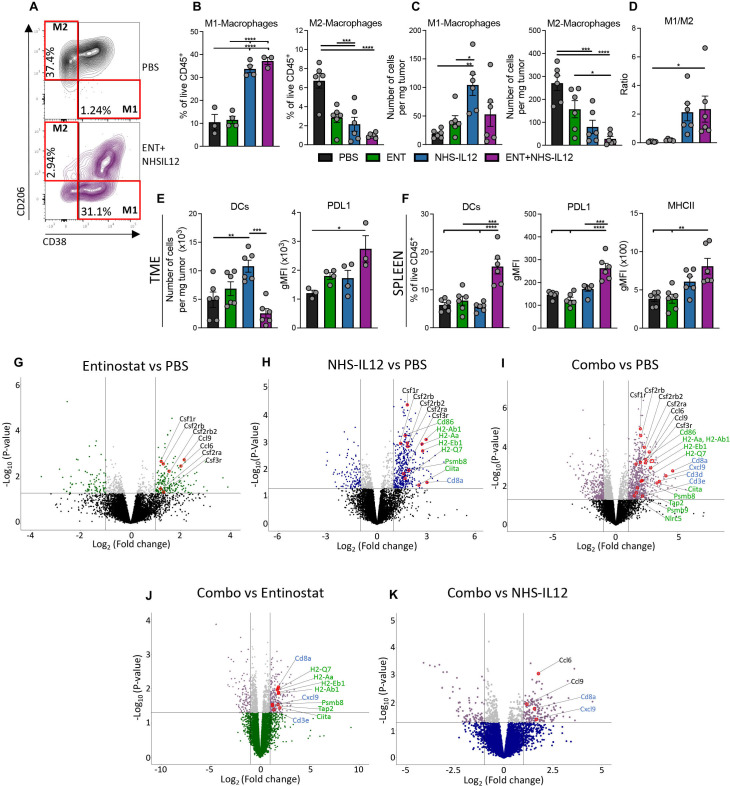
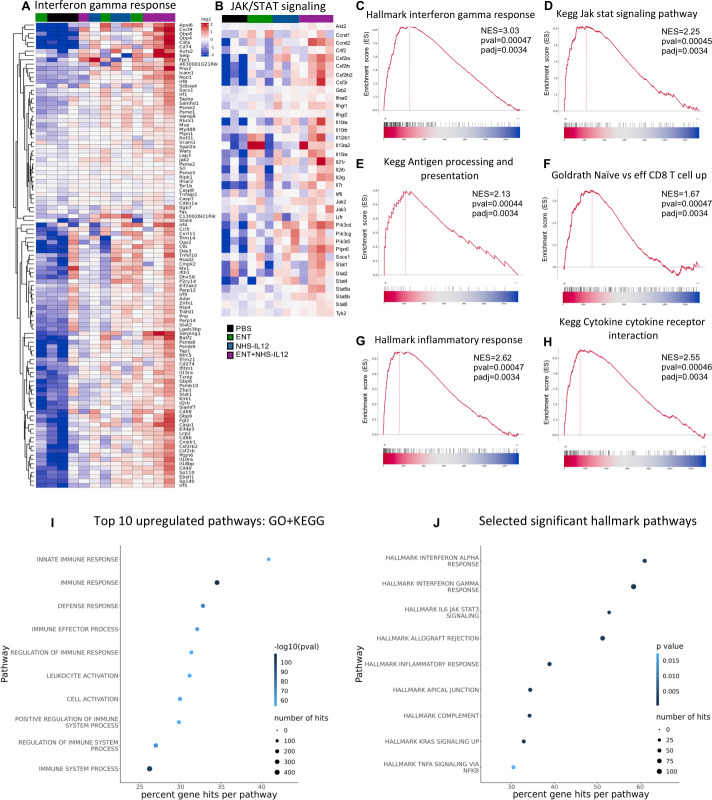
Background: Immune checkpoint blockade (ICB) has achieved unprecedented success in treating multiple cancer types. However, clinical benefit remains modest for most patients with solid malignancies due to primary or acquired resistance. Tumor-intrinsic loss of major histocompatibility complex class I (MHC-I) and aberrations in antigen processing machinery (APM) and interferon gamma (IFN-γ) pathways have been shown to play an important role in ICB resistance. While a plethora of combination treatments are being investigated to overcome ICB resistance, there are few identified preclinical models of solid tumors harboring these deficiencies to explore therapeutic interventions that can bypass ICB resistance. Here, we investigated the combination of the epigenetic modulator entinostat and the tumor-targeted immunocytokine NHS-IL12 in three different murine tumor models resistant to αPD-1/αPD-L1 (anti-programmed cell death protein 1/anti-programmed death ligand 1) and harboring MHC-I, APM, and IFN-γ response deficiencies and differing tumor mutational burden (TMB).
Methods: Entinostat and NHS-IL12 were administered to mice bearing TC-1/a9 (lung, HPV16 E6/E7+), CMT.64 lung, or RVP3 sarcoma tumors. Antitumor efficacy and survival were monitored. Comprehensive tumor microenvironment (TME) and spleen analysis of immune cells, cytokines, and chemokines was performed. Additionally, whole transcriptomic analysis was carried out on TC-1/a9 tumors. Cancer Genome Atlas (TCGA) datasets were analyzed for translational relevance.
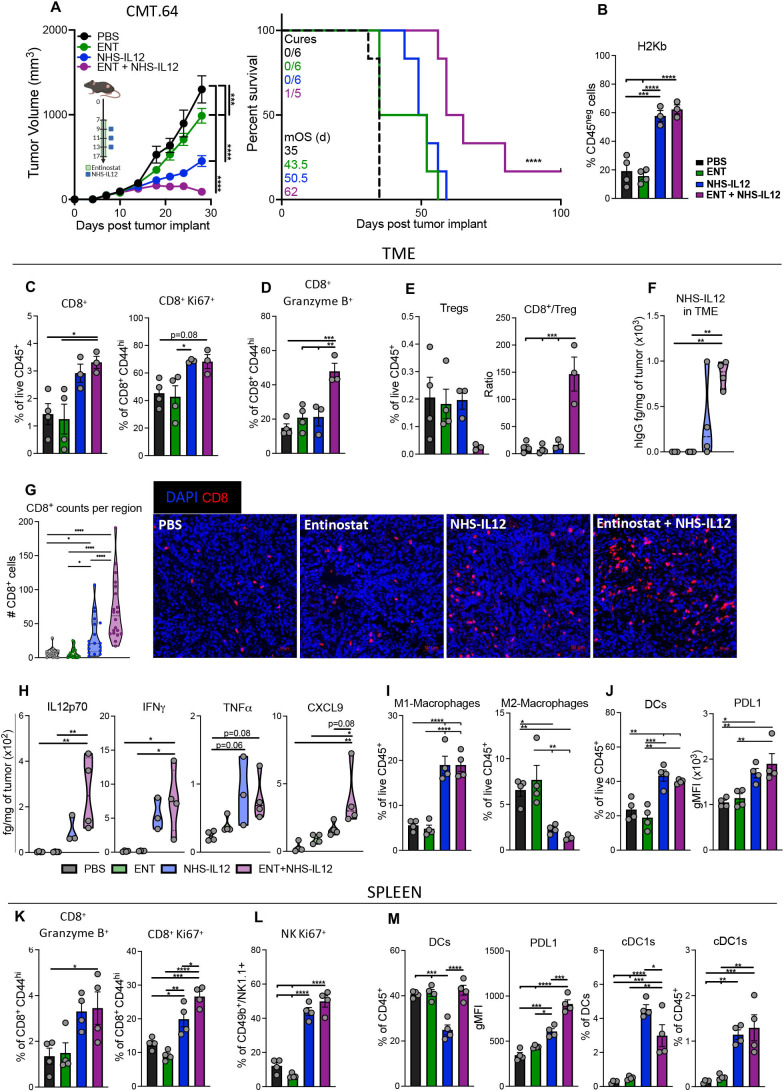
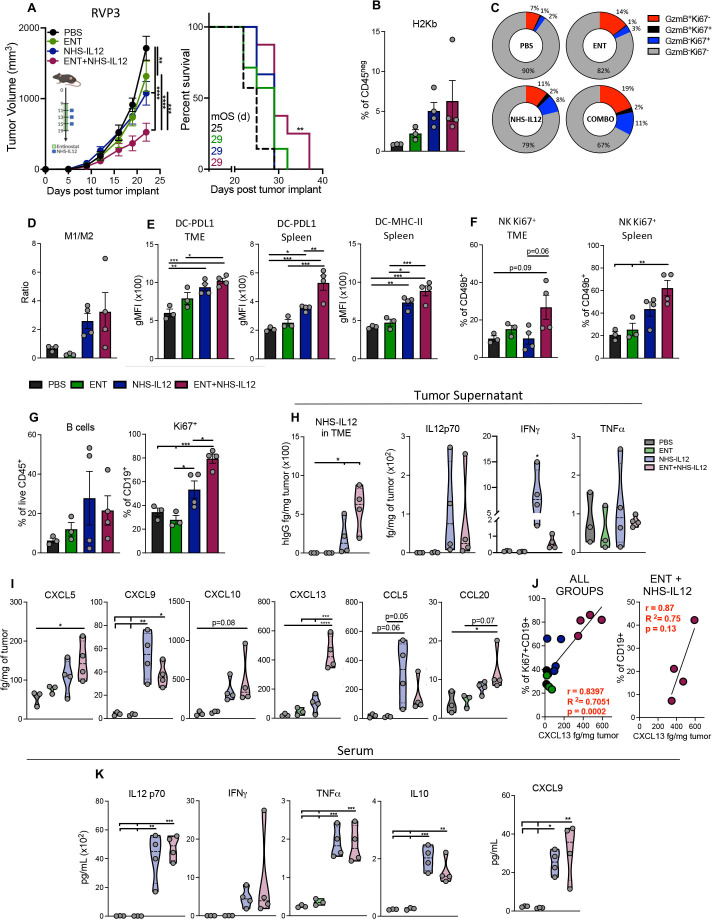
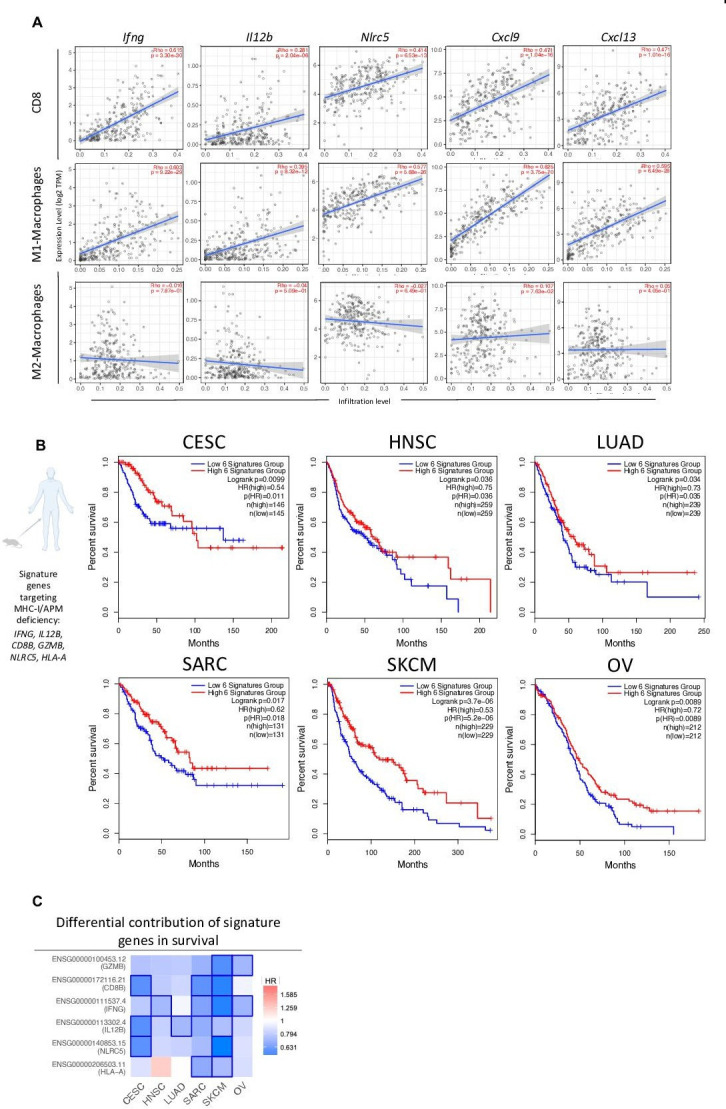
Results: We demonstrate that the combination of entinostat and NHS-IL12 therapy elicits potent antitumor activity and survival benefit through prolonged activation and tumor infiltration of cytotoxic CD8+ T cells, across αPD-1/αPD-L1 refractory tumors irrespective of TMB, including in the IFN-γ signaling-impaired RVP3 tumor model. The combination therapy promoted M1-like macrophages and activated antigen-presenting cells while decreasing M2-like macrophages and regulatory T cells in a tumor-dependent manner. This was associated with increased levels of IFN-γ, IL-12, chemokine (C-X-C motif) ligand 9 (CXCL9), and CXCL13 in the TME. Further, the combination therapy synergized to promote MHC-I and APM upregulation, and enrichment of JAK/STAT (janus kinase/signal transducers and activators of transcription), IFN-γ-response and antigen processing-associated pathways. A biomarker signature of the mechanism involved in these studies is associated with patients' overall survival across multiple tumor types.
Conclusions: Our findings provide a rationale for combining the tumor-targeting NHS-IL12 with the histone deacetylase inhibitor entinostat in the clinical setting for patients unresponsive to αPD-1/αPD-L1 and/or with innate deficiencies in tumor MHC-I, APM expression, and IFN-γ signaling.
Keywords: combined modality therapy; cytokines; immunotherapy; programmed cell death 1 receptor; tumor microenvironment.
© Author(s) (or their employer(s)) 2022. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: The Laboratory of Tumor Immunology and Biology/Center for Immuno-Oncology, NCI, has a Cooperative Research and Development Agreement with Syndax Pharmaceuticals and EMD Serono. KCH is now an AstraZeneca employee.
Figures
References
-
- Marabelle A, Fakih M, Lopez J, et al. . Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol 2020;21:1353–65. 10.1016/S1470-2045(20)30445-9 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials