

Learning representations of chromatin contacts using a recurrent neural network identifies genomic drivers of conformation
- PMID: 35764630
- PMCID: PMC9240038
- DOI: 10.1038/s41467-022-31337-w
Learning representations of chromatin contacts using a recurrent neural network identifies genomic drivers of conformation
Abstract
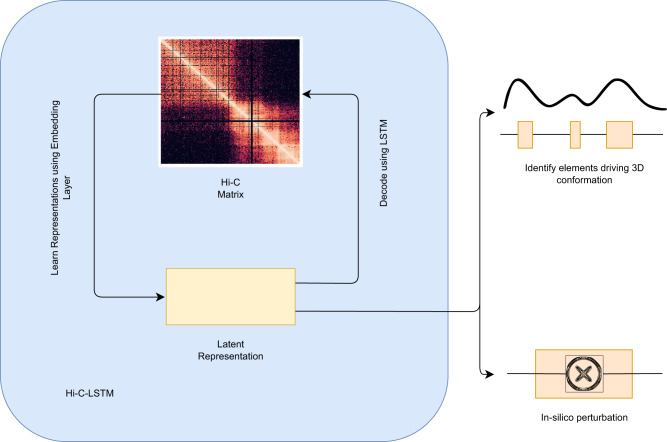
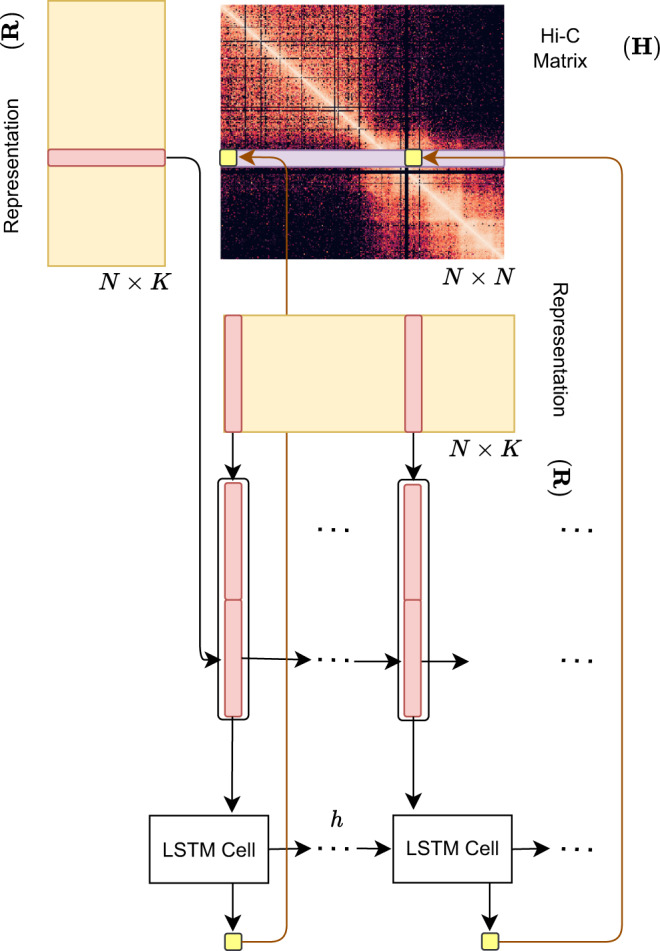
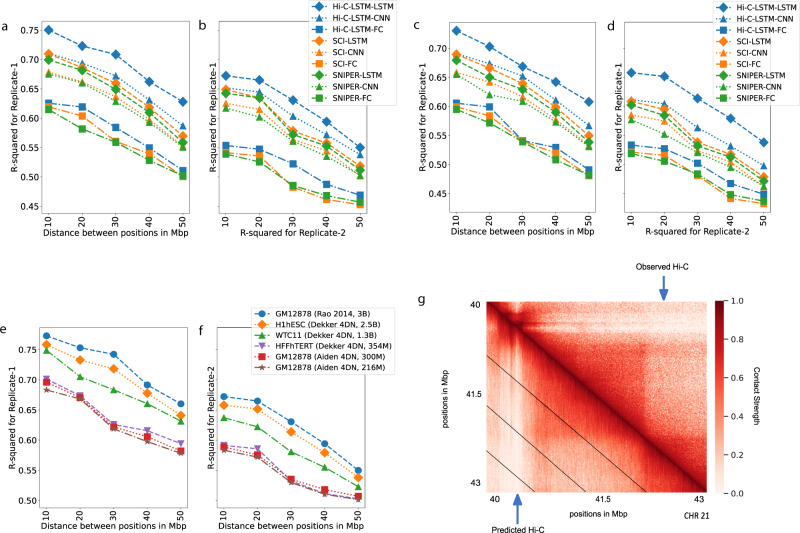
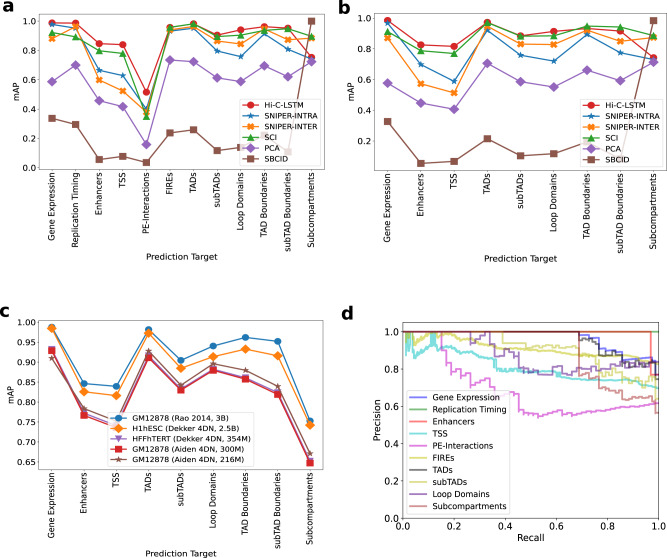
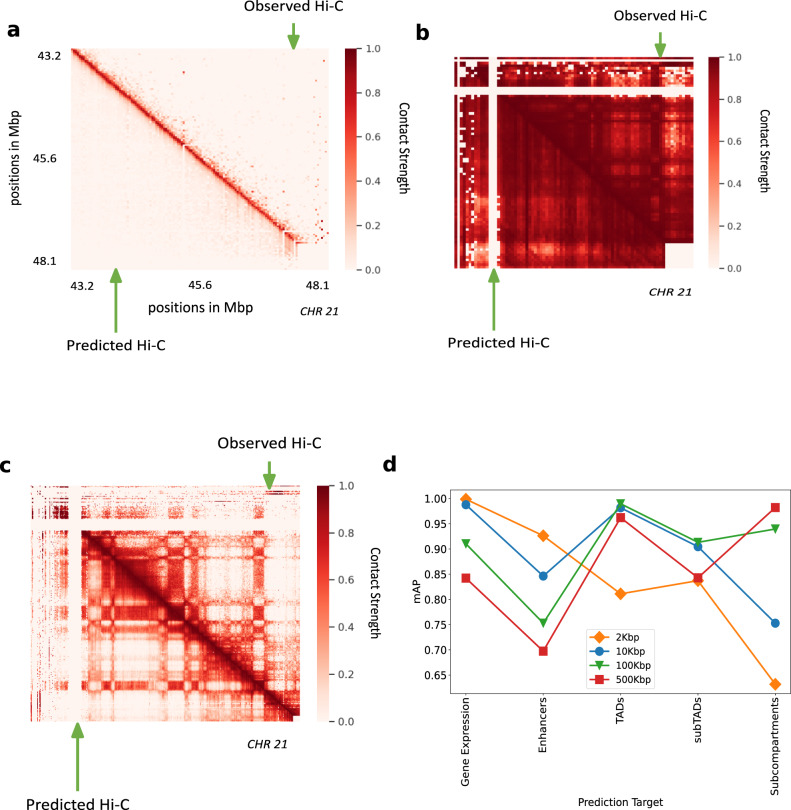
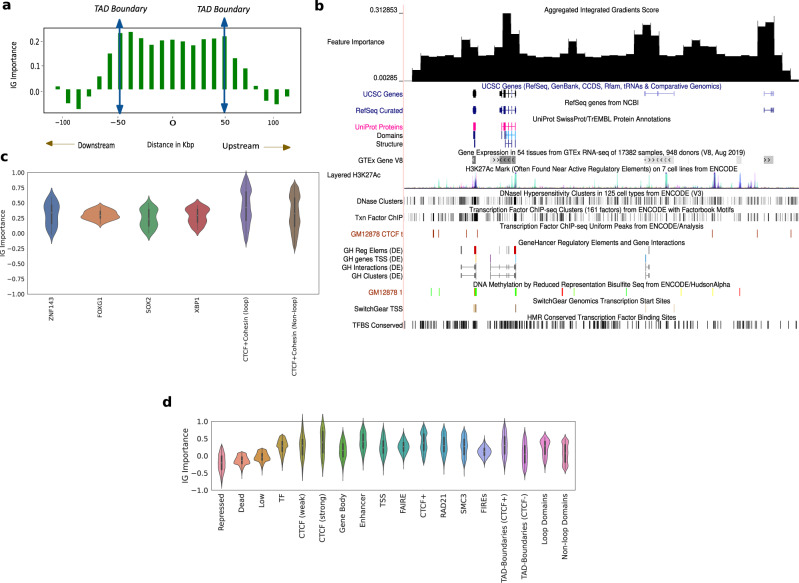
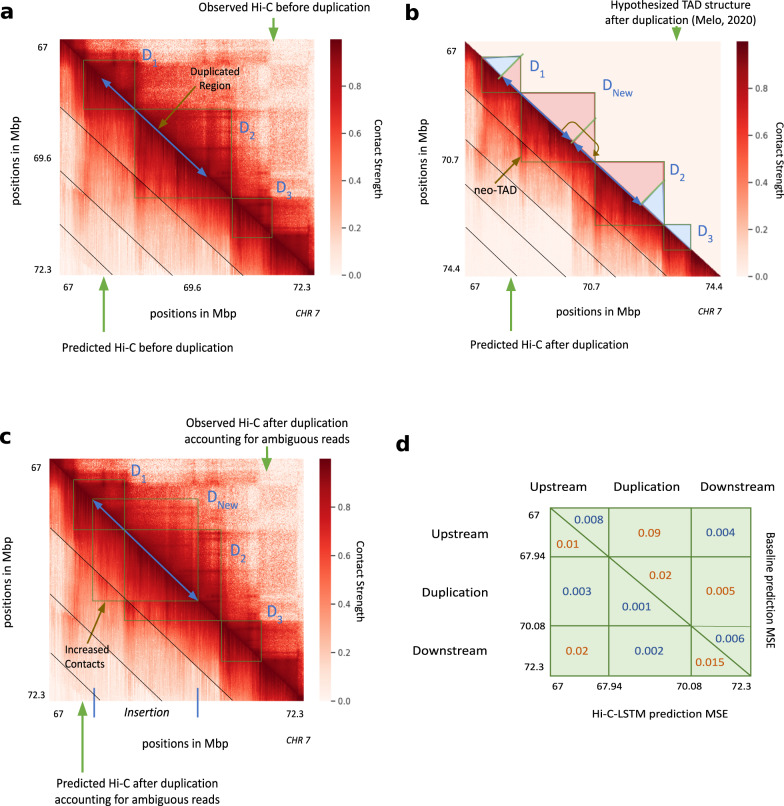
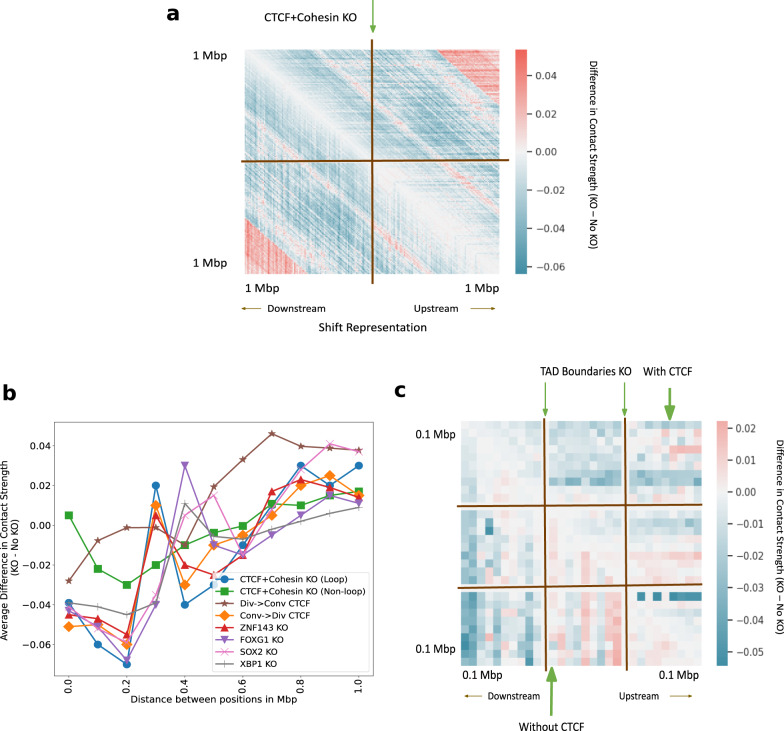
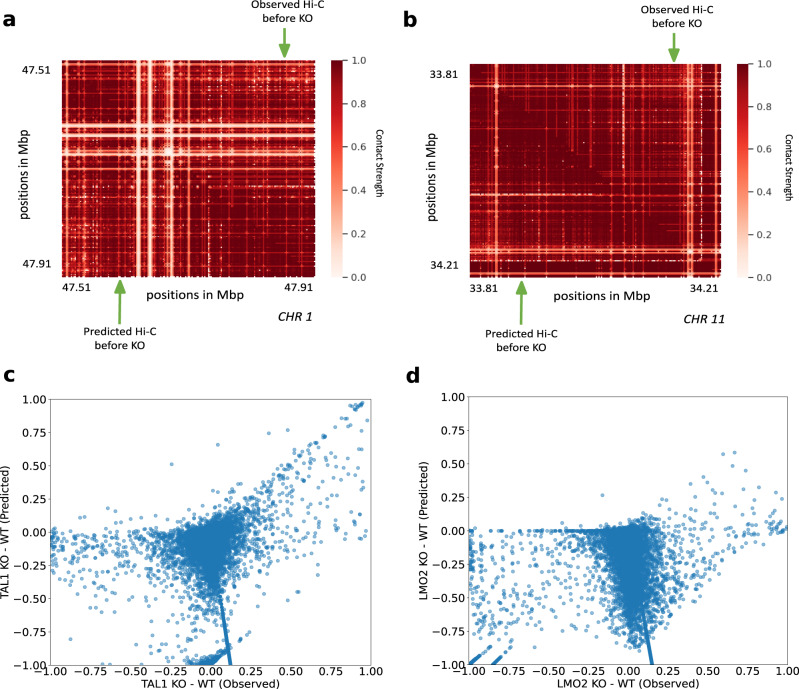
Despite the availability of chromatin conformation capture experiments, discerning the relationship between the 1D genome and 3D conformation remains a challenge, which limits our understanding of their affect on gene expression and disease. We propose Hi-C-LSTM, a method that produces low-dimensional latent representations that summarize intra-chromosomal Hi-C contacts via a recurrent long short-term memory neural network model. We find that these representations contain all the information needed to recreate the observed Hi-C matrix with high accuracy, outperforming existing methods. These representations enable the identification of a variety of conformation-defining genomic elements, including nuclear compartments and conformation-related transcription factors. They furthermore enable in-silico perturbation experiments that measure the influence of cis-regulatory elements on conformation.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Bengio Y, Courville A, Vincent P. Representation learning: A review and new perspectives. IEEE Trans. Pattern Anal. Mach. Intell. 2013;35:1798–828. - PubMed
-
- Seide, F., Li, G. & Yu, D. Conversational speech transcription using context-dependent deep neural networks. In Proc. 12th Annual Conference of theInternational Speech Communication Association. 430–440 (2011).
-
- Boulanger-Lewandowski, N., Bengio, Y. & Vincent, P. Modeling temporal dependencies in high-dimensional sequences: application to polyphonic music generation and transcription. Preprint at arXiv:1206.6392 (2012).
-
- Krizhevsky A, Sutskever I, Hinton GE. Imagenet classification with deep convolutional neural networks. Commun. ACM. 2017;60:84–90.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
