

Cvm1 is a component of multiple vacuolar contact sites required for sphingolipid homeostasis
- PMID: 35766971
- PMCID: PMC9247719
- DOI: 10.1083/jcb.202103048
Cvm1 is a component of multiple vacuolar contact sites required for sphingolipid homeostasis
Abstract
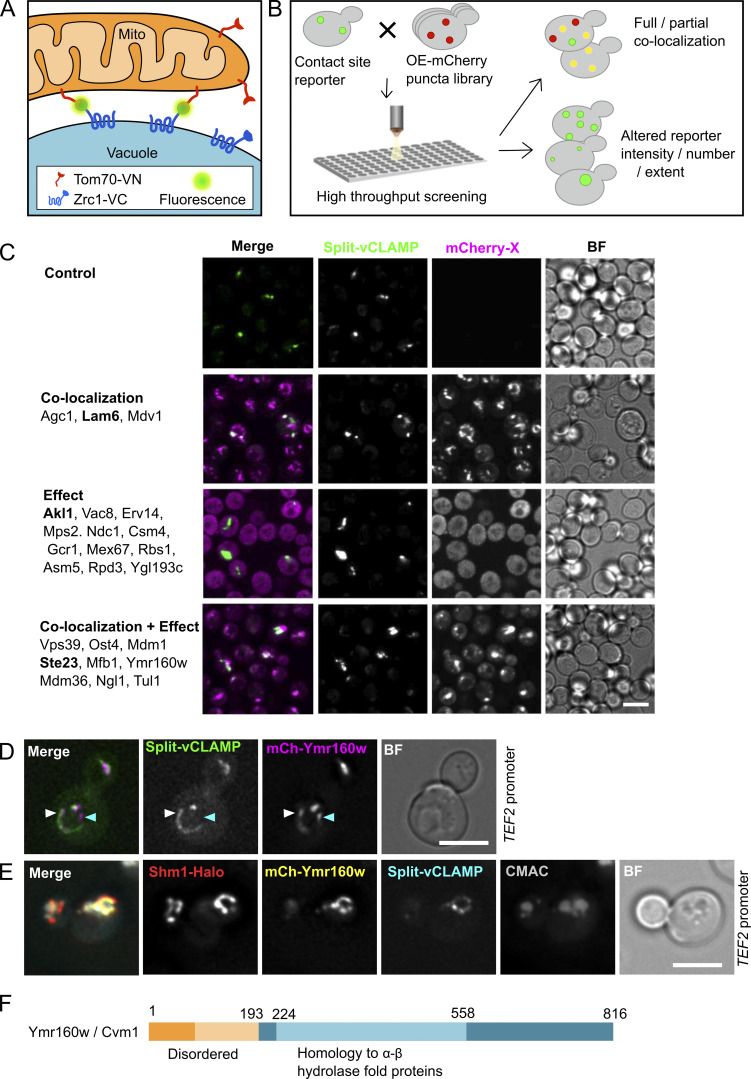
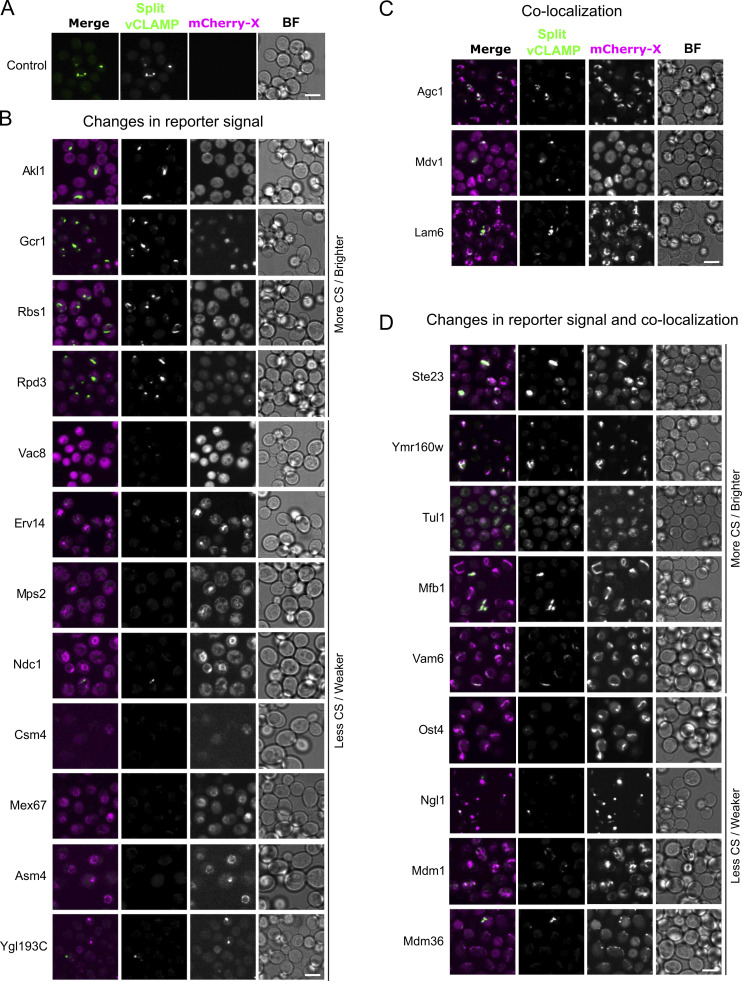
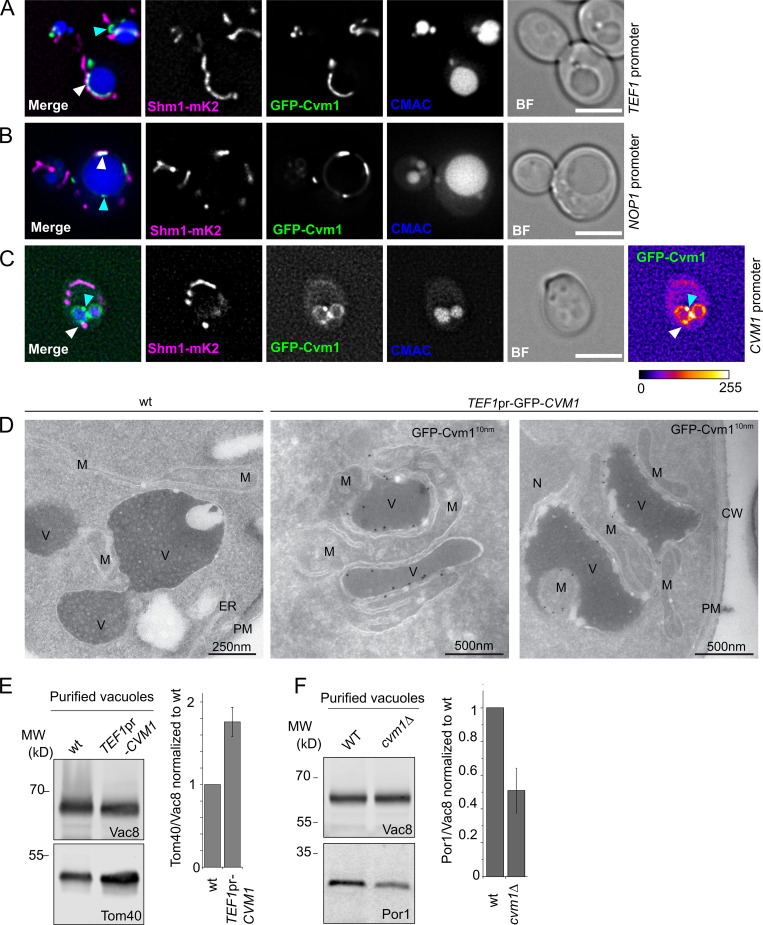
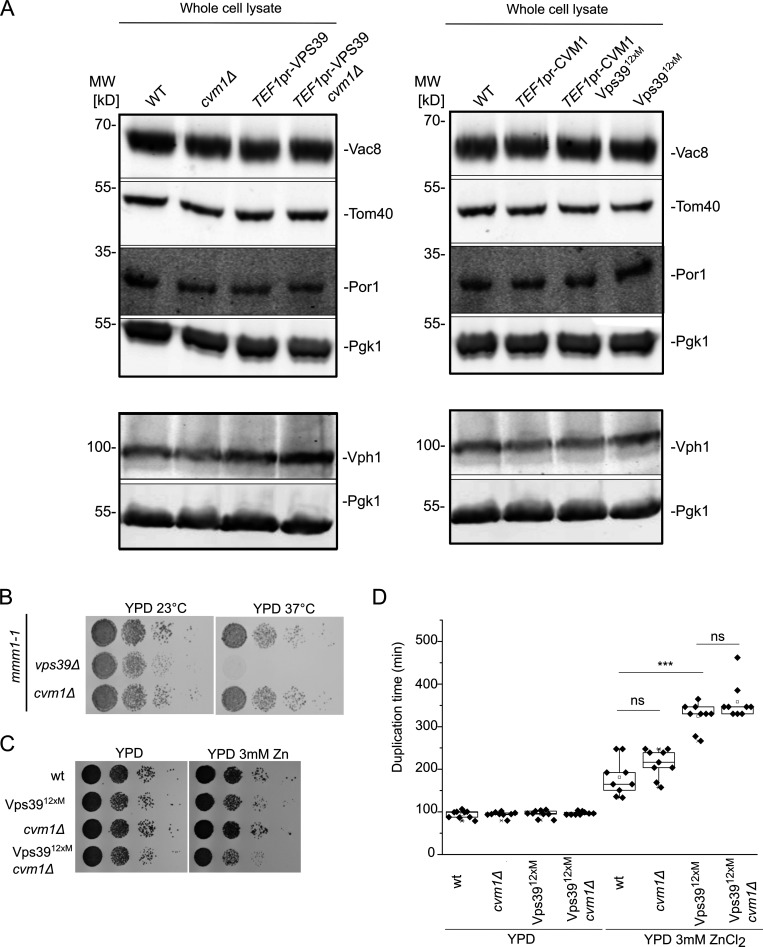
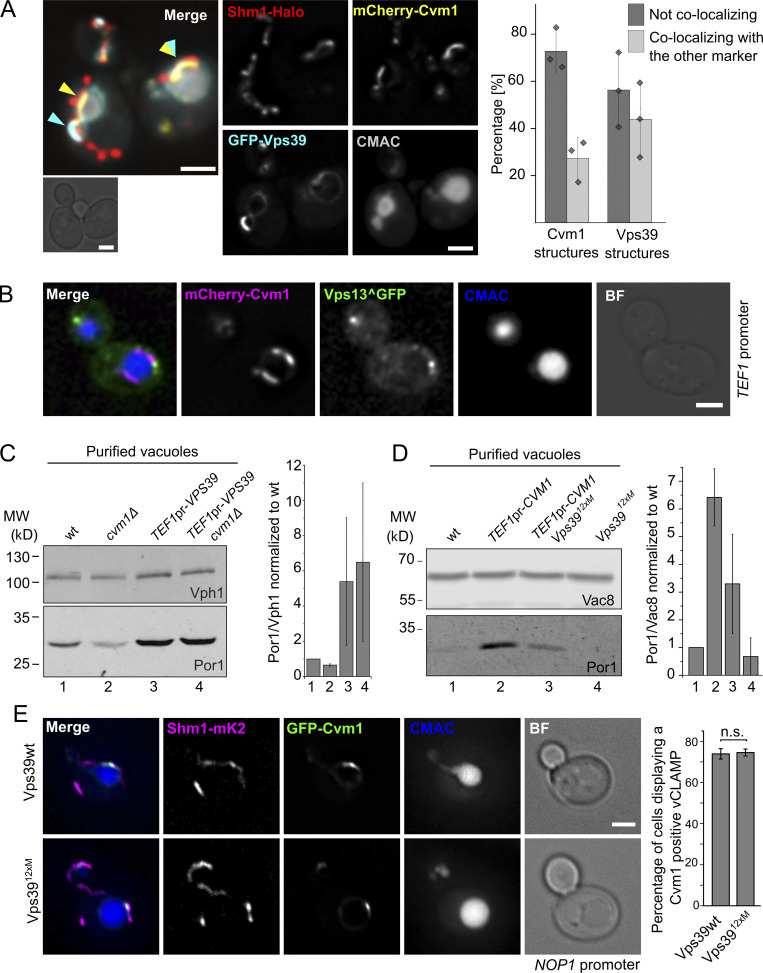
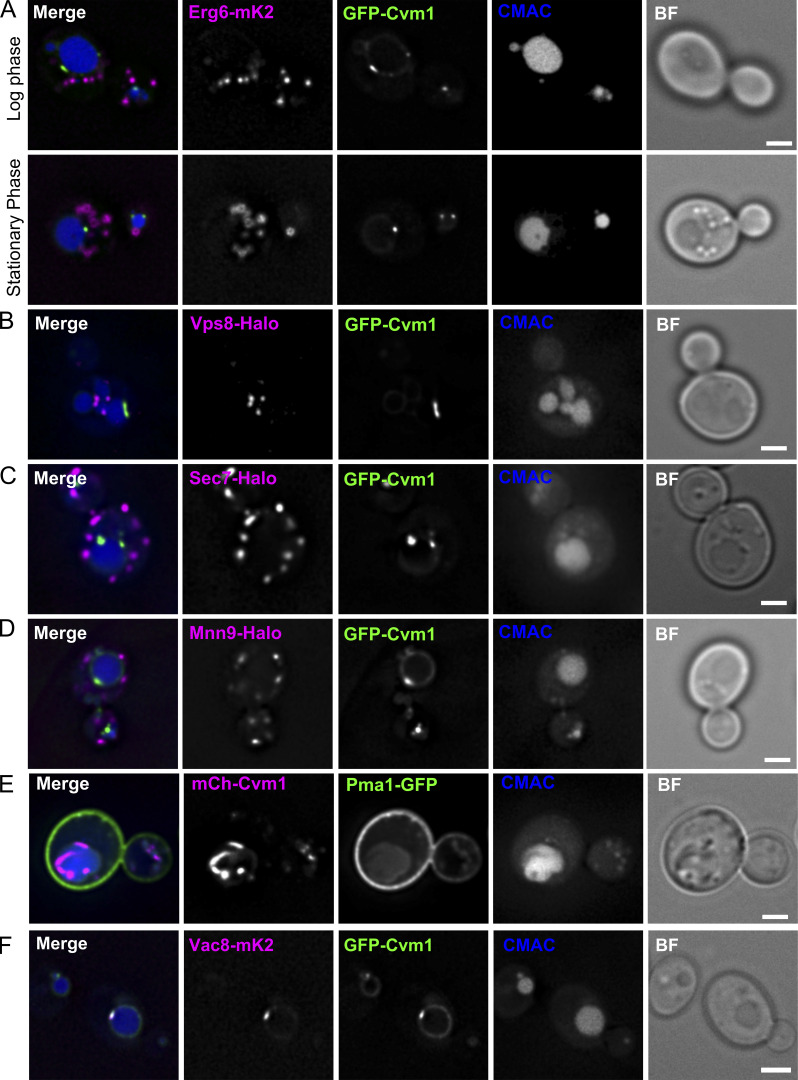
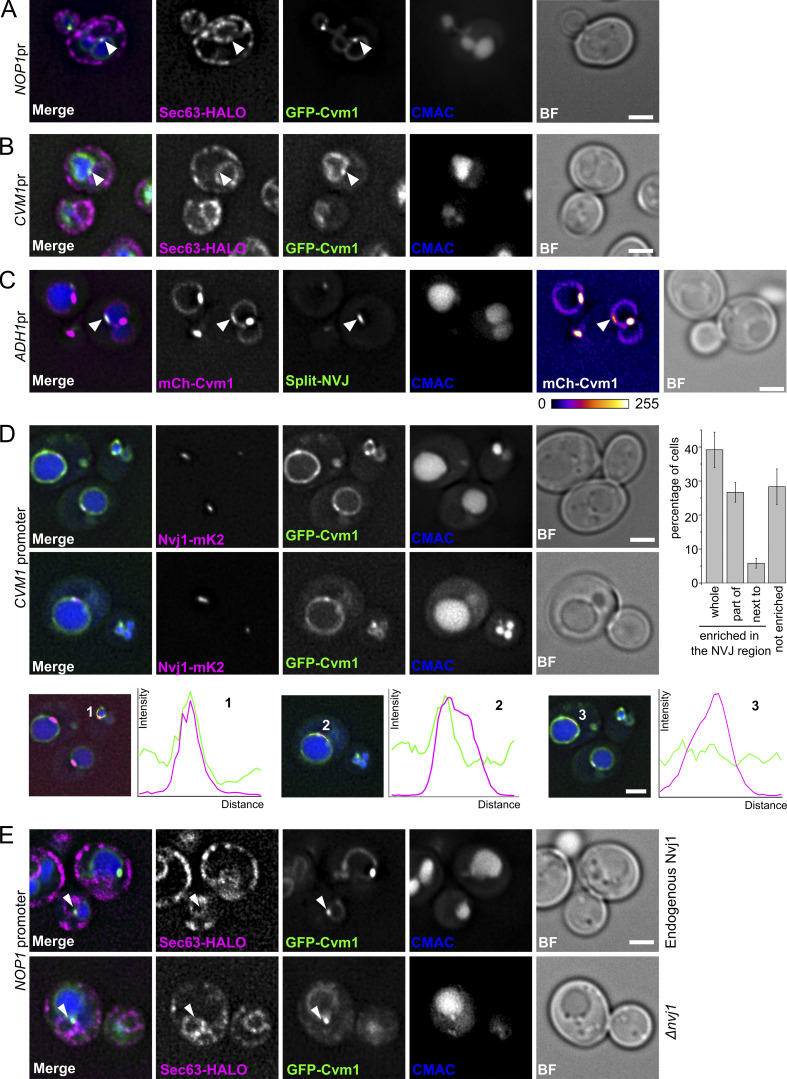
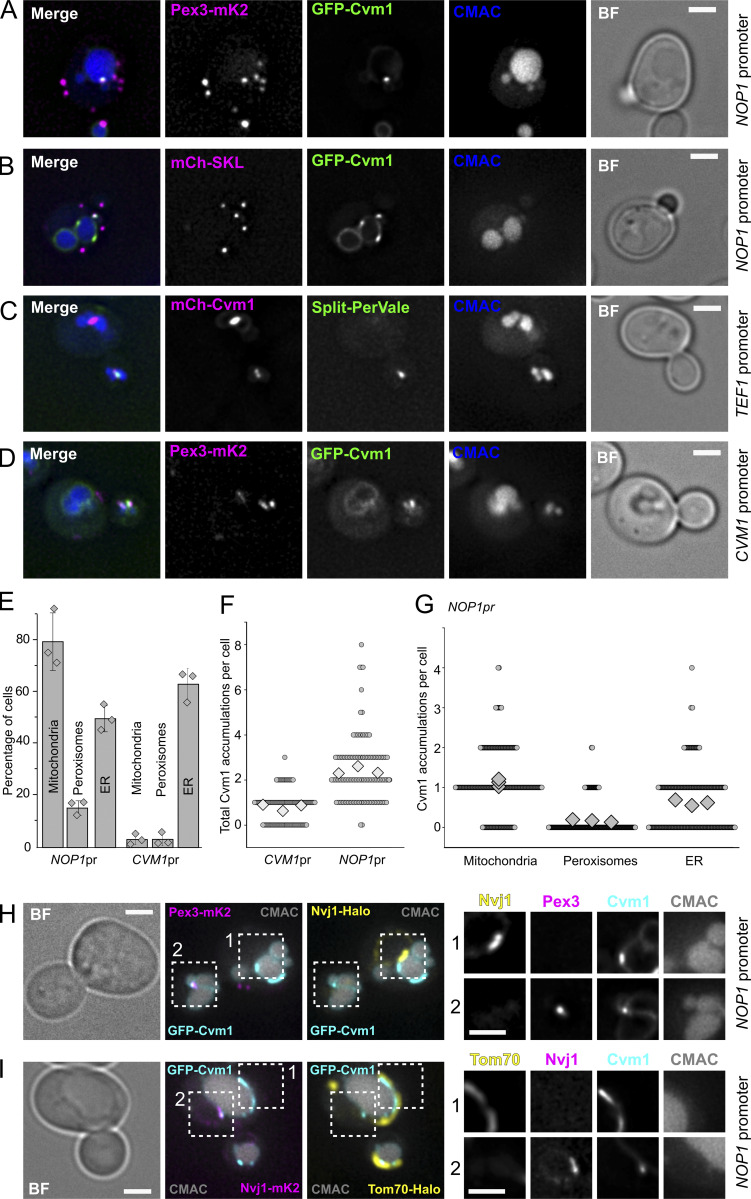
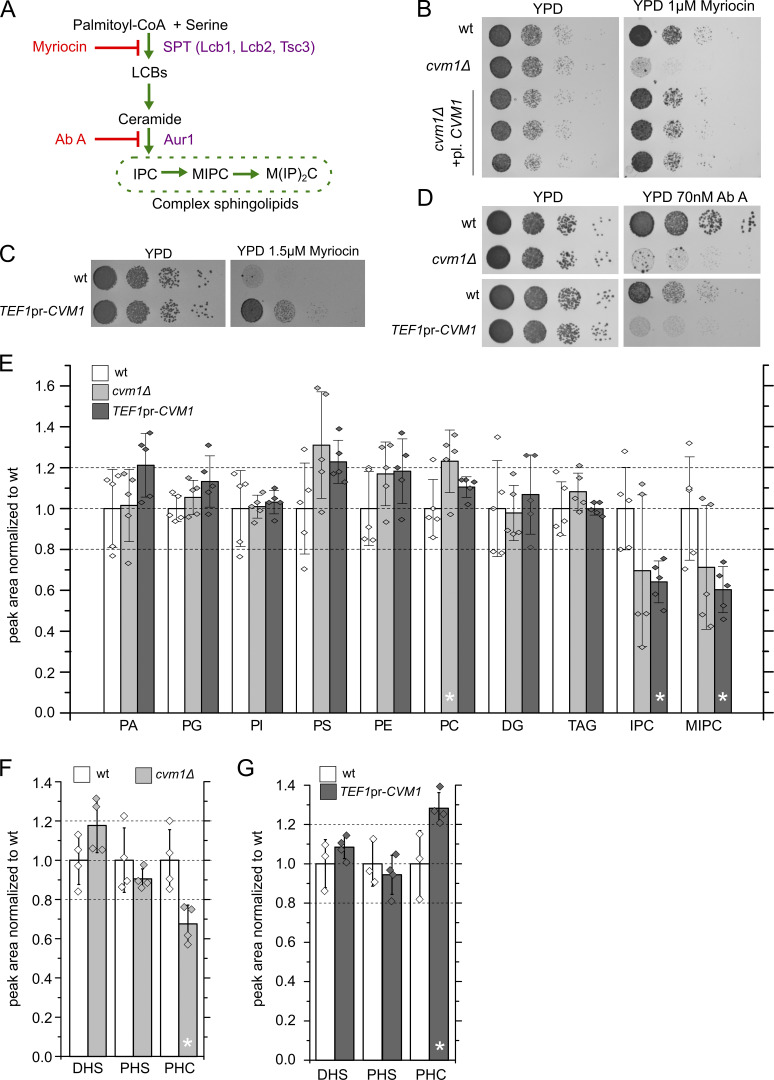
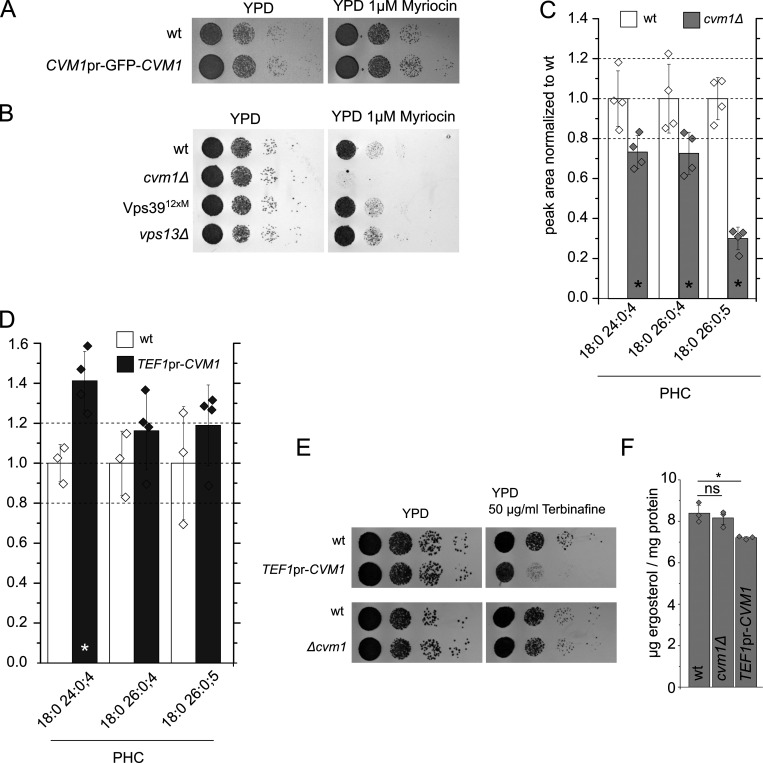
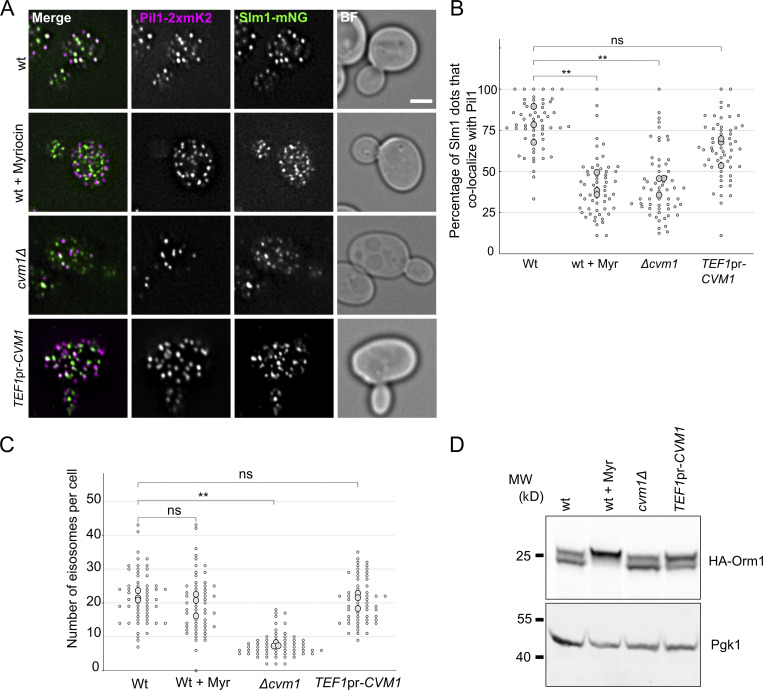
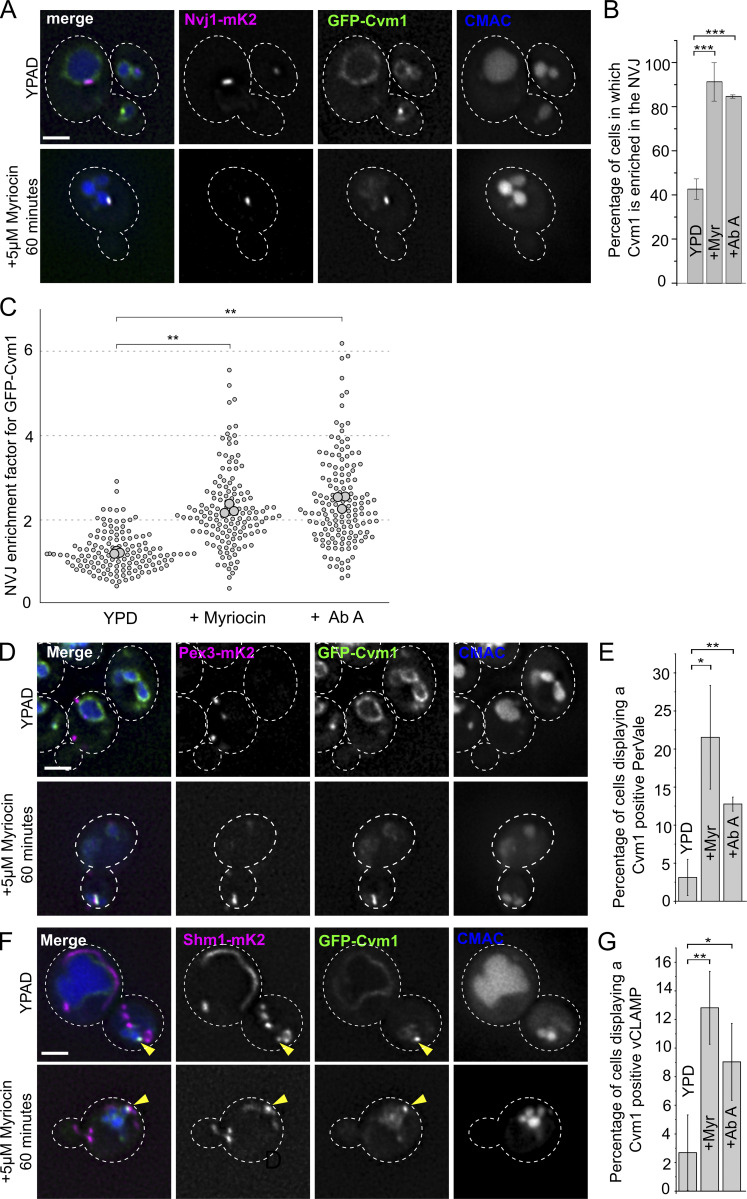
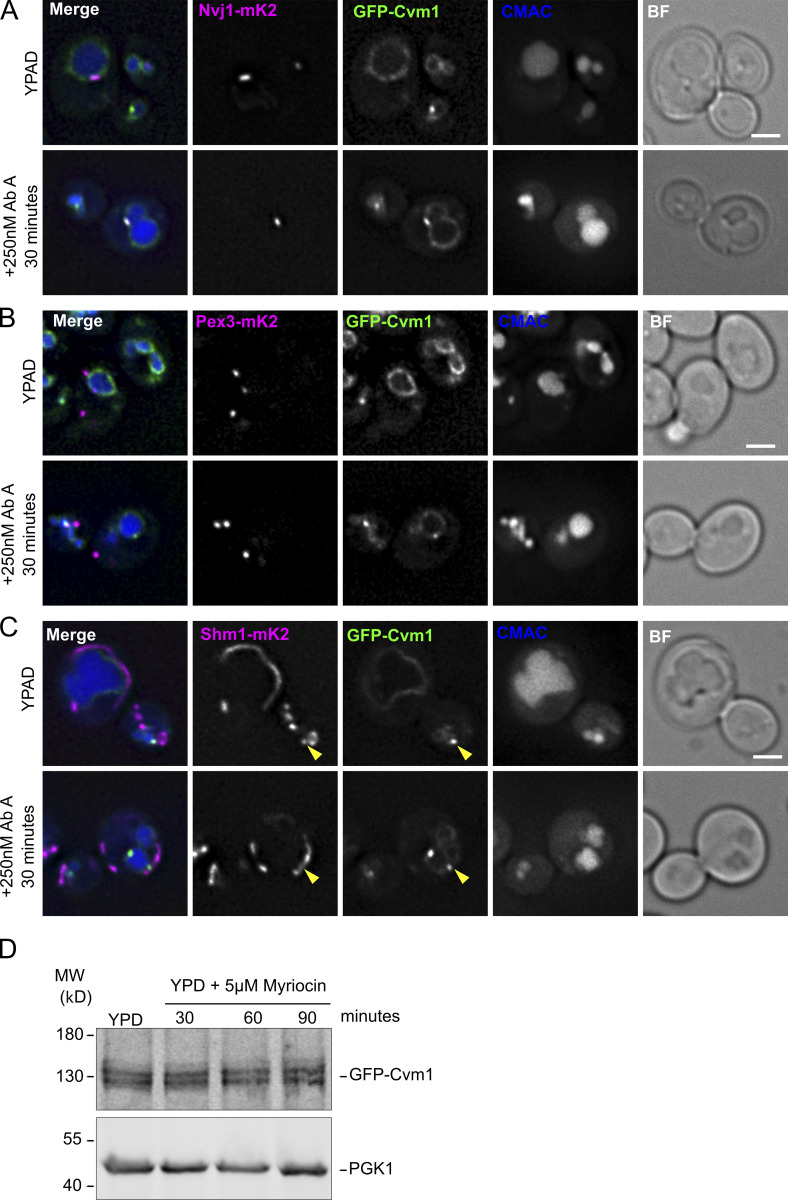
Membrane contact sites are specialized platforms formed between most organelles that enable them to exchange metabolites and influence the dynamics of each other. The yeast vacuole is a degradative organelle equivalent to the lysosome in higher eukaryotes with important roles in ion homeostasis and metabolism. Using a high-content microscopy screen, we identified Ymr160w (Cvm1, for contact of the vacuole membrane 1) as a novel component of three different contact sites of the vacuole: with the nuclear endoplasmic reticulum, the mitochondria, and the peroxisomes. At the vacuole-mitochondria contact site, Cvm1 acts as a tether independently of previously known tethers. We show that changes in Cvm1 levels affect sphingolipid homeostasis, altering the levels of multiple sphingolipid classes and the response of sphingolipid-sensing signaling pathways. Furthermore, the contact sites formed by Cvm1 are induced upon a decrease in sphingolipid levels. Altogether, our work identifies a novel protein that forms multiple contact sites and supports a role of lysosomal contacts in sphingolipid homeostasis.
© 2022 Bisinski et al.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
